Wu Fengbo, Xu Ting, He Gu, Ouyang Liang, Han Bo, Peng Cheng, Song Xiangrong, Xiang Mingli
Department of Pharmacy and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, China.
Int J Mol Sci. 2012 Nov 23;13(12):15668-78. doi: 10.3390/ijms131215668.
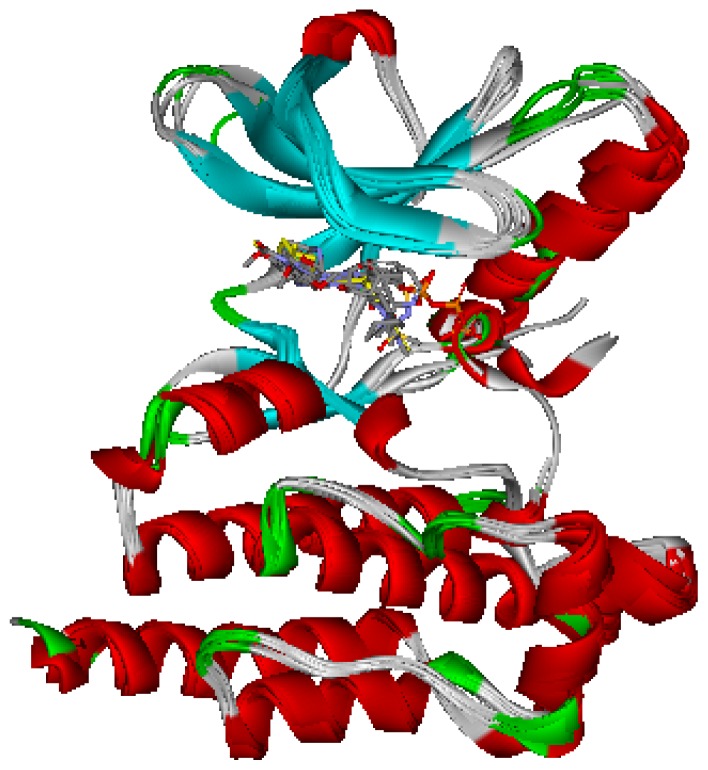
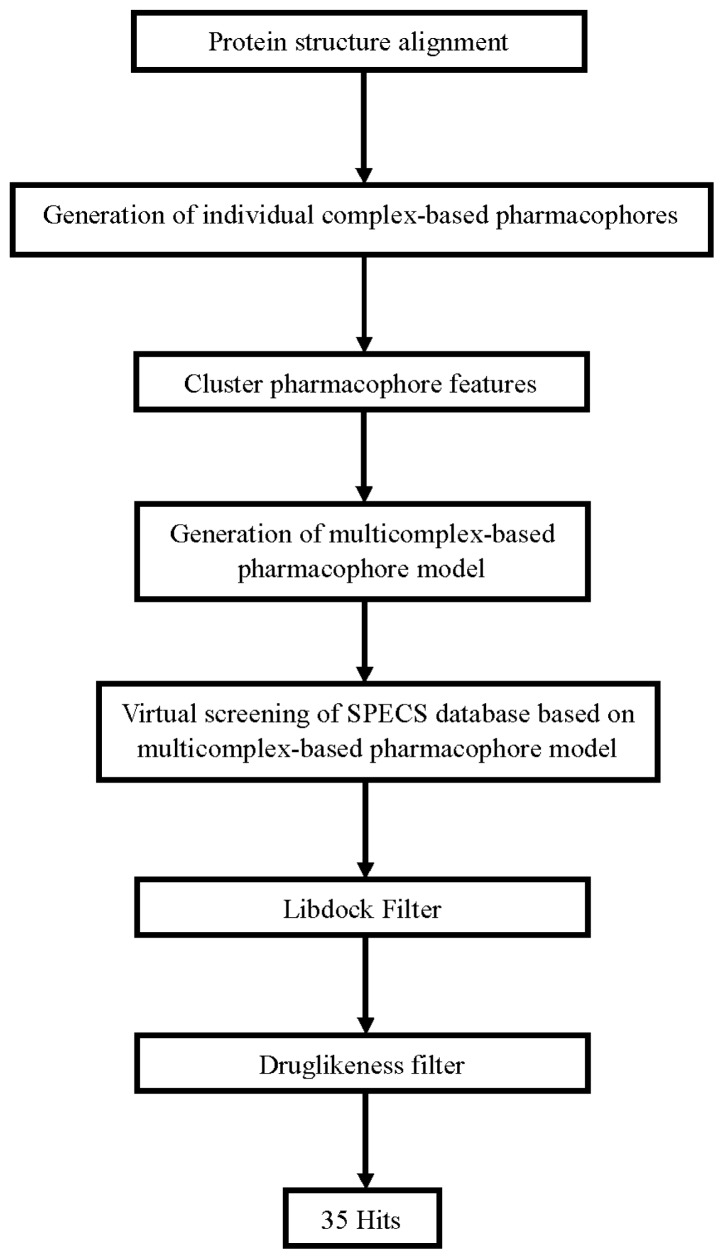
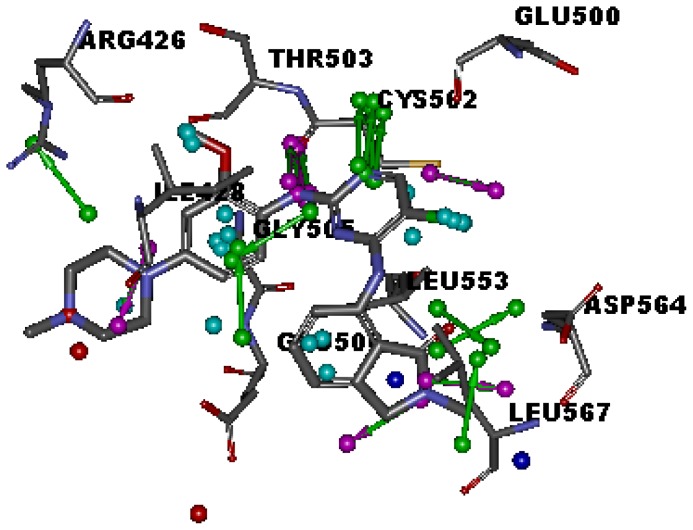
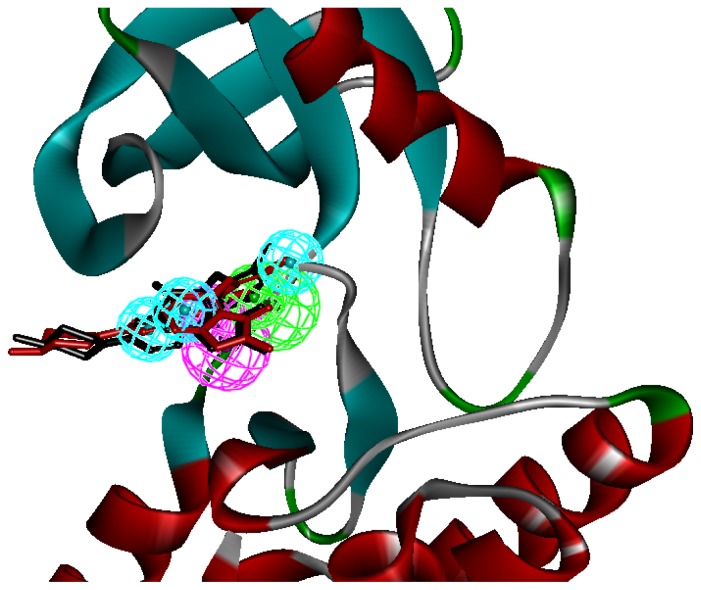
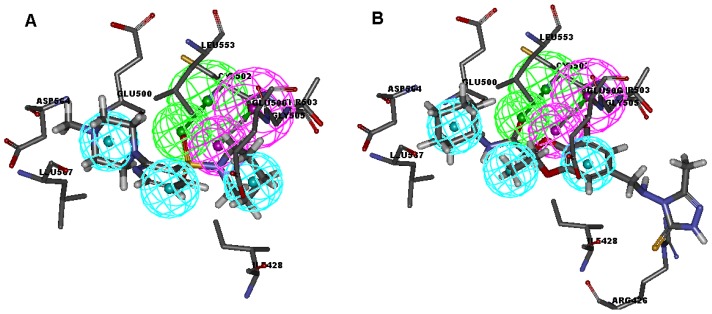
Focal adhesion kinase (FAK) is a tyrosine kinase that functions as a key orchestrator of signals leading to invasion and metastasis. In the current study, the multicomplex-based pharmacophore (MCBP)-guided method has been suggested to generate a comprehensive pharmacophore of FAK kinase based on seven crystal structures of FAK-inhibitor complexes. In this investigation, a hybrid protocol of virtual screening methods, comprising of pharmacophore model-based virtual screening (PB-VS) and docking-based virtual screening (DB-VS), is used for retrieving new FAK inhibitors from commercially available chemical databases. This hybrid virtual screening approach was then applied to screen several chemical databases, including the Specs (202,408 compounds) database. Thirty-five compounds were selected from the final hits and should be shifted to experimental studies. These results may provide important information for further research of novel FAK inhibitors.
粘着斑激酶(FAK)是一种酪氨酸激酶,在导致侵袭和转移的信号传导中起关键协调作用。在本研究中,基于多复合物的药效团(MCBP)引导方法被用于根据FAK抑制剂复合物的七个晶体结构生成FAK激酶的综合药效团。在这项研究中,一种虚拟筛选方法的混合方案,包括基于药效团模型的虚拟筛选(PB-VS)和基于对接的虚拟筛选(DB-VS),被用于从商业化学数据库中检索新的FAK抑制剂。然后,这种混合虚拟筛选方法被应用于筛选几个化学数据库,包括Specs(202,408种化合物)数据库。从最终命中物中选择了35种化合物,并应转向实验研究。这些结果可能为新型FAK抑制剂的进一步研究提供重要信息。