Laboratory of DNA Information Analysis, Human Genome Center, Institute of Medical Science, The University of Tokyo, 4-6-1, Shirokanedai, Minato-ku, Tokyo 108-8639, Japan.
Nucleic Acids Res. 2013 Apr;41(7):e89. doi: 10.1093/nar/gkt126. Epub 2013 Mar 6.
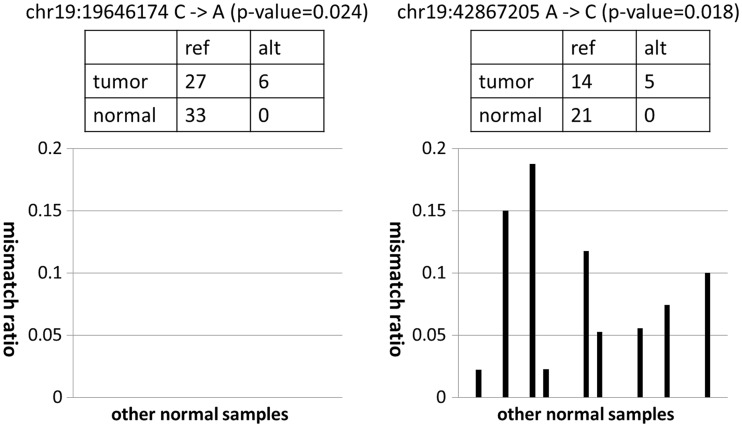
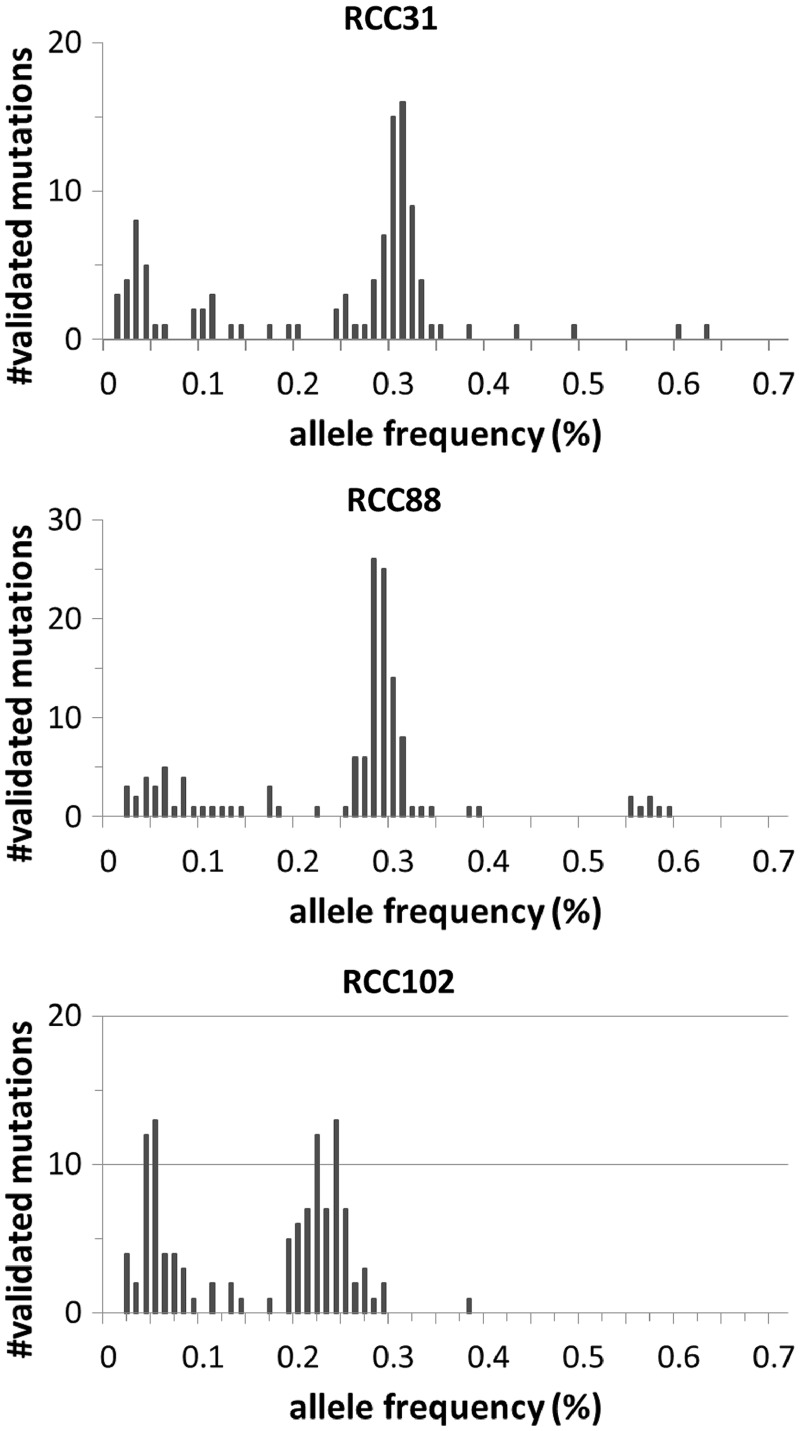
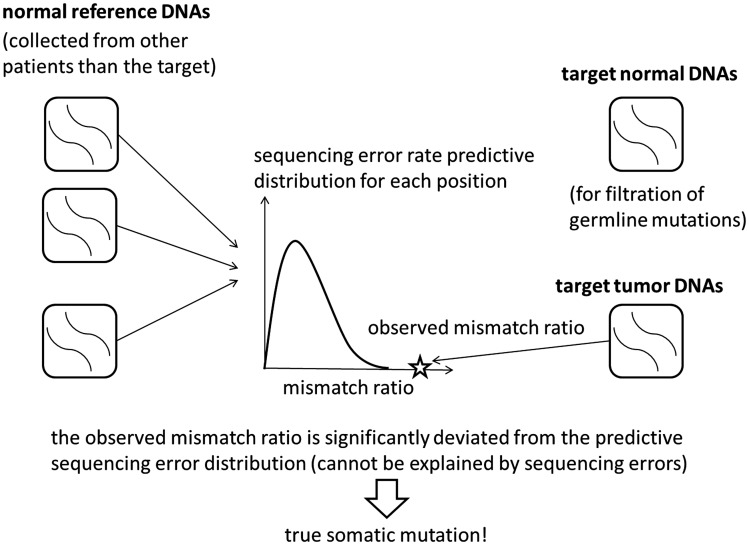
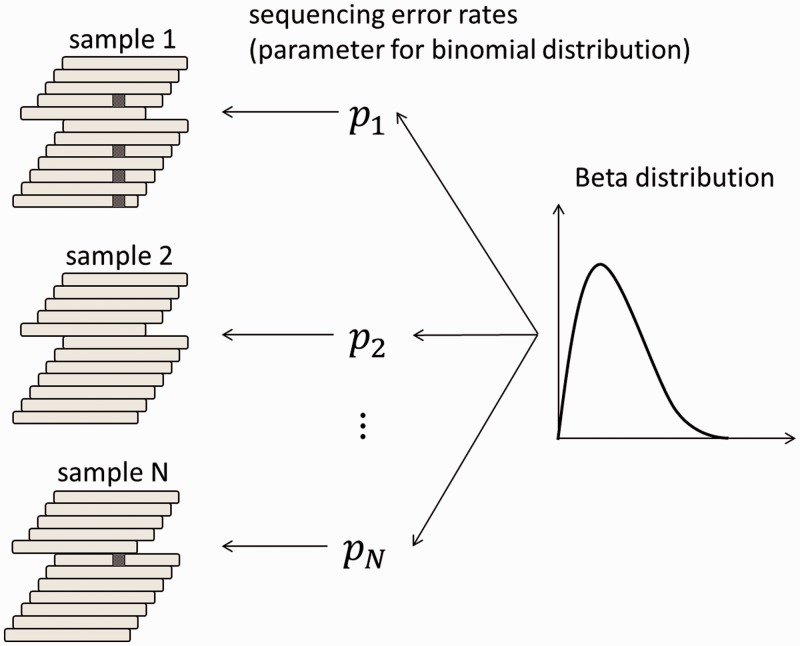
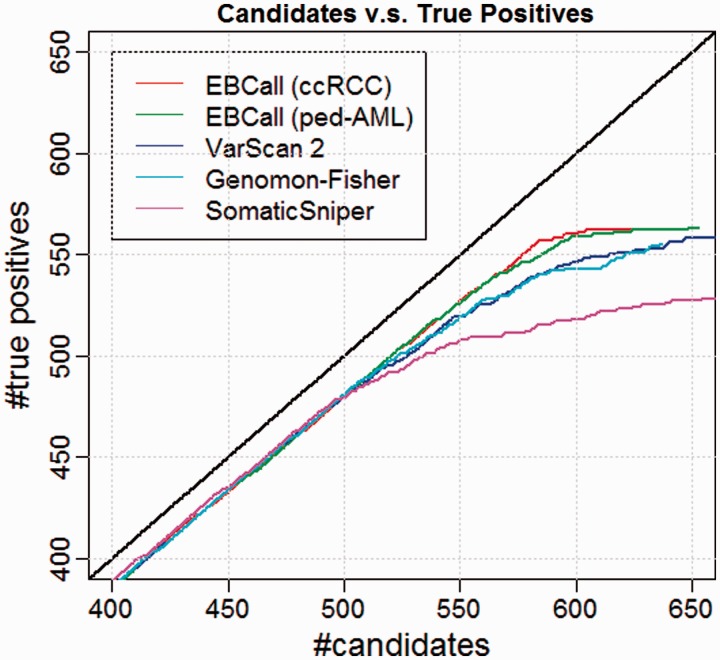
Recent advances in high-throughput sequencing technologies have enabled a comprehensive dissection of the cancer genome clarifying a large number of somatic mutations in a wide variety of cancer types. A number of methods have been proposed for mutation calling based on a large amount of sequencing data, which is accomplished in most cases by statistically evaluating the difference in the observed allele frequencies of possible single nucleotide variants between tumours and paired normal samples. However, an accurate detection of mutations remains a challenge under low sequencing depths or tumour contents. To overcome this problem, we propose a novel method, Empirical Bayesian mutation Calling (https://github.com/friend1ws/EBCall), for detecting somatic mutations. Unlike previous methods, the proposed method discriminates somatic mutations from sequencing errors based on an empirical Bayesian framework, where the model parameters are estimated using sequencing data from multiple non-paired normal samples. Using 13 whole-exome sequencing data with 87.5-206.3 mean sequencing depths, we demonstrate that our method not only outperforms several existing methods in the calling of mutations with moderate allele frequencies but also enables accurate calling of mutations with low allele frequencies (≤ 10%) harboured within a minor tumour subpopulation, thus allowing for the deciphering of fine substructures within a tumour specimen.
高通量测序技术的最新进展使人们能够全面解析癌症基因组,阐明了多种癌症类型中大量的体细胞突变。已经提出了许多基于大量测序数据进行突变调用的方法,这些方法在大多数情况下通过统计评估肿瘤和配对正常样本之间可能的单核苷酸变异的观察等位基因频率的差异来完成。然而,在测序深度低或肿瘤含量低的情况下,准确检测突变仍然是一个挑战。为了解决这个问题,我们提出了一种新的方法,经验贝叶斯突变调用(https://github.com/friend1ws/EBCall),用于检测体细胞突变。与以前的方法不同,该方法基于经验贝叶斯框架从测序错误中区分体细胞突变,其中使用来自多个非配对正常样本的测序数据来估计模型参数。使用 13 个全外显子测序数据,平均测序深度为 87.5-206.3,我们证明我们的方法不仅在调用具有中等等位基因频率的突变方面优于几种现有方法,而且还能够准确调用低等位基因频率(≤10%)的突变,这些突变存在于小肿瘤亚群中,从而允许对肿瘤标本中的精细亚结构进行解析。