Division of Genetics and Cell Biology, Dulbecco Telethon Institute at Dibit, San Raffaele Scientific Institute, Milan, Italy.
Nat Med. 2013 Apr;19(4):488-93. doi: 10.1038/nm.3092. Epub 2013 Mar 24.
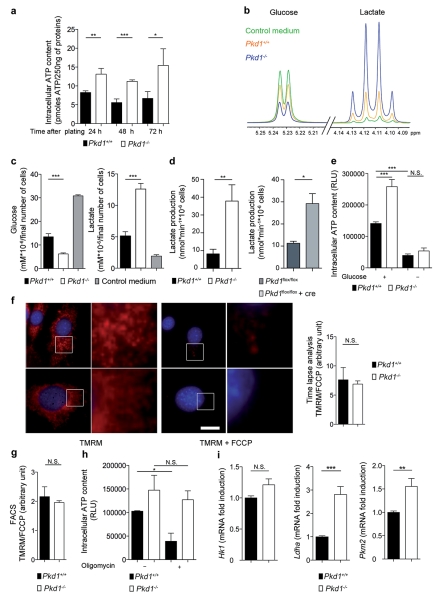
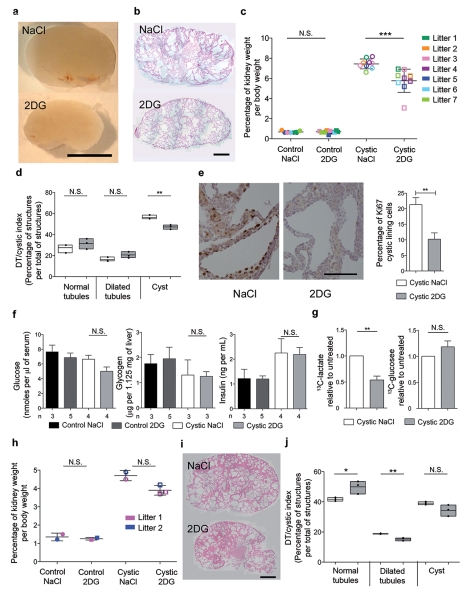
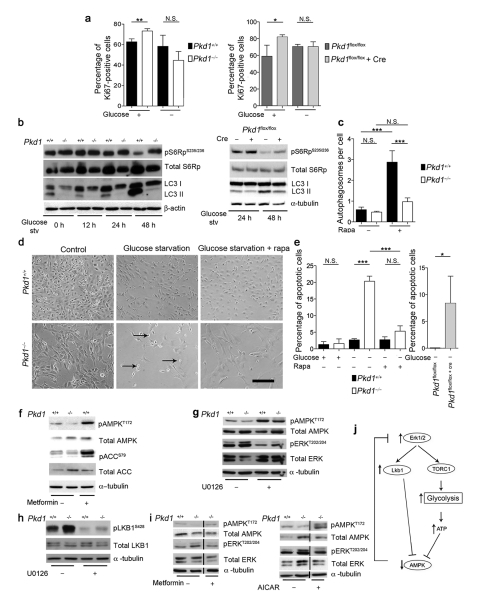
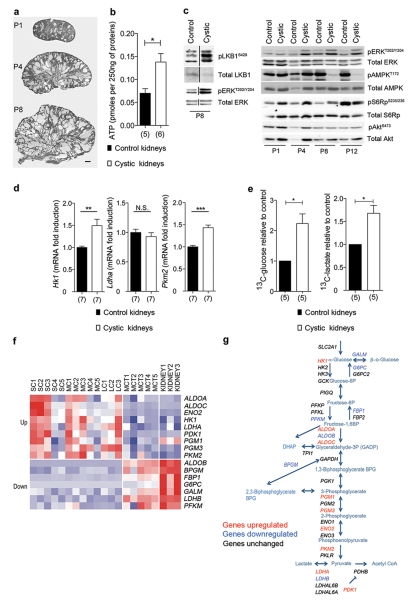
Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic disorder characterized by bilateral renal cyst formation. Recent identification of signaling cascades deregulated in ADPKD has led to the initiation of several clinical trials, but an approved therapy is still lacking. Using a metabolomic approach, we identify a pathogenic pathway in this disease that can be safely targeted for therapy. We show that mutation of PKD1 results in enhanced glycolysis in cells in a mouse model of PKD and in kidneys from humans with ADPKD. Glucose deprivation resulted in lower proliferation and higher apoptotic rates in PKD1-mutant cells than in nondeprived cells. Notably, two distinct PKD mouse models treated with 2-deoxyglucose (2DG), to inhibit glycolysis, had lower kidney weight, volume, cystic index and proliferation rates as compared to nontreated mice. These metabolic alterations depend on the extracellular signal-related kinase (ERK) pathway acting in a dual manner by inhibiting the liver kinase B1 (LKB1)-AMP-activated protein kinase (AMPK) axis on the one hand while activating the mTOR complex 1 (mTORC1)-glycolytic cascade on the other. Enhanced metabolic rates further inhibit AMPK. Forced activation of AMPK acts in a negative feedback loop, restoring normal ERK activity. Taken together, these data indicate that defective glucose metabolism is intimately involved in the pathobiology of ADPKD. Our findings provide a strong rationale for a new therapeutic strategy using existing drugs, either individually or in combination.
常染色体显性多囊肾病(ADPKD)是一种常见的遗传性疾病,其特征为双侧肾脏囊肿形成。最近发现 ADPKD 中信号级联失调,这导致了几项临床试验的启动,但仍缺乏批准的治疗方法。我们使用代谢组学方法,在该疾病中确定了一条可安全作为治疗靶点的致病途径。我们表明,PKD1 的突变导致 PKD 小鼠模型和 ADPKD 患者肾脏中的细胞糖酵解增强。与未剥夺葡萄糖的细胞相比,葡萄糖剥夺导致 PKD1 突变细胞的增殖率降低和凋亡率升高。值得注意的是,用 2-脱氧葡萄糖(2DG)抑制糖酵解治疗两种不同的 PKD 小鼠模型,与未治疗的小鼠相比,肾脏重量、体积、囊泡指数和增殖率均降低。这些代谢变化取决于细胞外信号调节激酶(ERK)通路,一方面通过抑制肝激酶 B1(LKB1)-AMP 激活蛋白激酶(AMPK)轴,另一方面激活 mTOR 复合物 1(mTORC1)-糖酵解级联,从而发挥双重作用。增强的代谢率进一步抑制 AMPK。AMPK 的强制激活在负反馈回路中起作用,恢复正常的 ERK 活性。综上所述,这些数据表明,葡萄糖代谢缺陷与 ADPKD 的病理生物学密切相关。我们的发现为使用现有药物单独或联合使用提供了一种新的治疗策略的强有力依据。