Rogers Kelly A, Moreno Sarah E, Smith Laurie A, Husson Hervé, Bukanov Nikolay O, Ledbetter Steven R, Budman Yeva, Lu Yuefeng, Wang Bing, Ibraghimov-Beskrovnaya Oxana, Natoli Thomas A
Department of Rare Renal Disease Research, Sanofi-Genzyme R&D Center, Framingham, Massachusetts.
Department of Analytical Research and Development, Sanofi Corporation, Waltham, Massachusetts.
Physiol Rep. 2016 Jun;4(12). doi: 10.14814/phy2.12846.
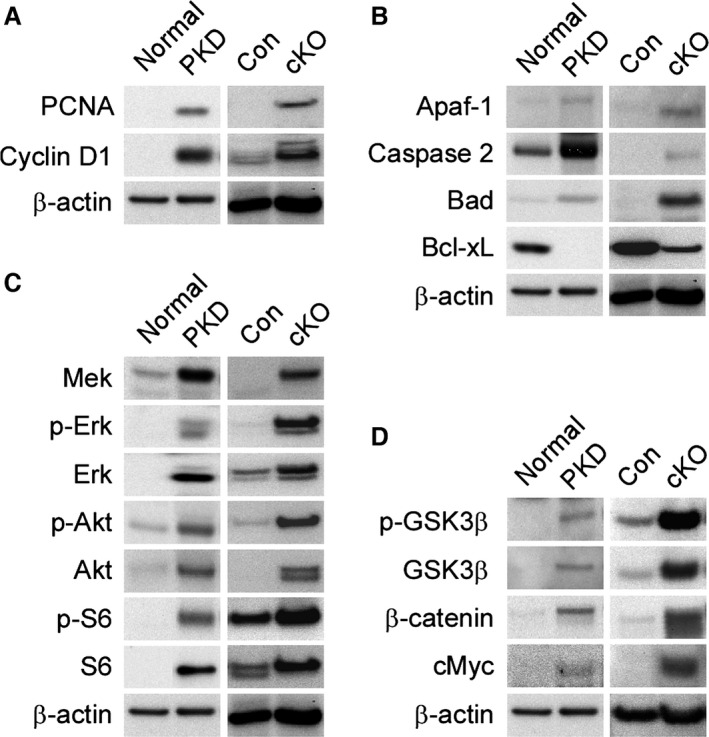
Development of a disease-modifying therapy to treat autosomal dominant polycystic kidney disease (ADPKD) requires well-characterized preclinical models that accurately reflect the pathology and biochemical changes associated with the disease. Using a Pkd1 conditional knockout mouse, we demonstrate that subtly altering the timing and extent of Pkd1 deletion can have a significant impact on the origin and severity of kidney cyst formation. Pkd1 deletion on postnatal day 1 or 2 results in cysts arising from both the cortical and medullary regions, whereas deletion on postnatal days 3-8 results in primarily medullary cyst formation. Altering the extent of Pkd1 deletion by modulating the tamoxifen dose produces dose-dependent changes in the severity, but not origin, of cystogenesis. Limited Pkd1 deletion produces progressive kidney cystogenesis, accompanied by interstitial fibrosis and loss of kidney function. Cyst growth occurs in two phases: an early, rapid growth phase, followed by a later, slow growth period. Analysis of biochemical pathway changes in cystic kidneys reveals dysregulation of the cell cycle, increased proliferation and apoptosis, activation of Mek-Erk, Akt-mTOR, and Wnt-β-catenin signaling pathways, and altered glycosphingolipid metabolism that resemble the biochemical changes occurring in human ADPKD kidneys. These pathways are normally active in neonatal mouse kidneys until repressed around 3 weeks of age; however, they remain active following Pkd1 deletion. Together, this work describes the key parameters to accurately model the pathological and biochemical changes associated with ADPKD in a conditional mouse model.
开发一种用于治疗常染色体显性多囊肾病(ADPKD)的疾病修饰疗法需要特征明确的临床前模型,该模型能准确反映与该疾病相关的病理和生化变化。利用Pkd1条件性敲除小鼠,我们证明,微妙地改变Pkd1缺失的时间和程度会对肾囊肿形成的起源和严重程度产生重大影响。出生后第1天或第2天敲除Pkd1会导致皮质和髓质区域都出现囊肿,而出生后第3 - 8天敲除则主要导致髓质囊肿形成。通过调节他莫昔芬剂量来改变Pkd1缺失的程度,会在囊肿形成的严重程度而非起源方面产生剂量依赖性变化。有限的Pkd1缺失会导致进行性肾囊肿形成,伴有间质纤维化和肾功能丧失。囊肿生长分两个阶段:早期的快速生长阶段,随后是后期的缓慢生长阶段。对囊性肾中生化途径变化的分析揭示了细胞周期失调、增殖和凋亡增加、Mek - Erk、Akt - mTOR和Wnt - β - 连环蛋白信号通路激活以及糖鞘脂代谢改变,这些变化类似于人类ADPKD肾中发生的生化变化。这些途径在新生小鼠肾中通常是活跃的,直到约3周龄时受到抑制;然而,在Pkd1缺失后它们仍保持活跃。总之,这项工作描述了在条件性小鼠模型中准确模拟与ADPKD相关的病理和生化变化的关键参数。