Department of Microbiology and Immunology, H.M. Bligh Cancer Research Laboratories, Chicago Medical School, Rosalind Franklin University of Medicine and Science, North Chicago, IL, USA.
Oncogenesis. 2012 Apr 2;1(4):e5. doi: 10.1038/oncsis.2012.5.
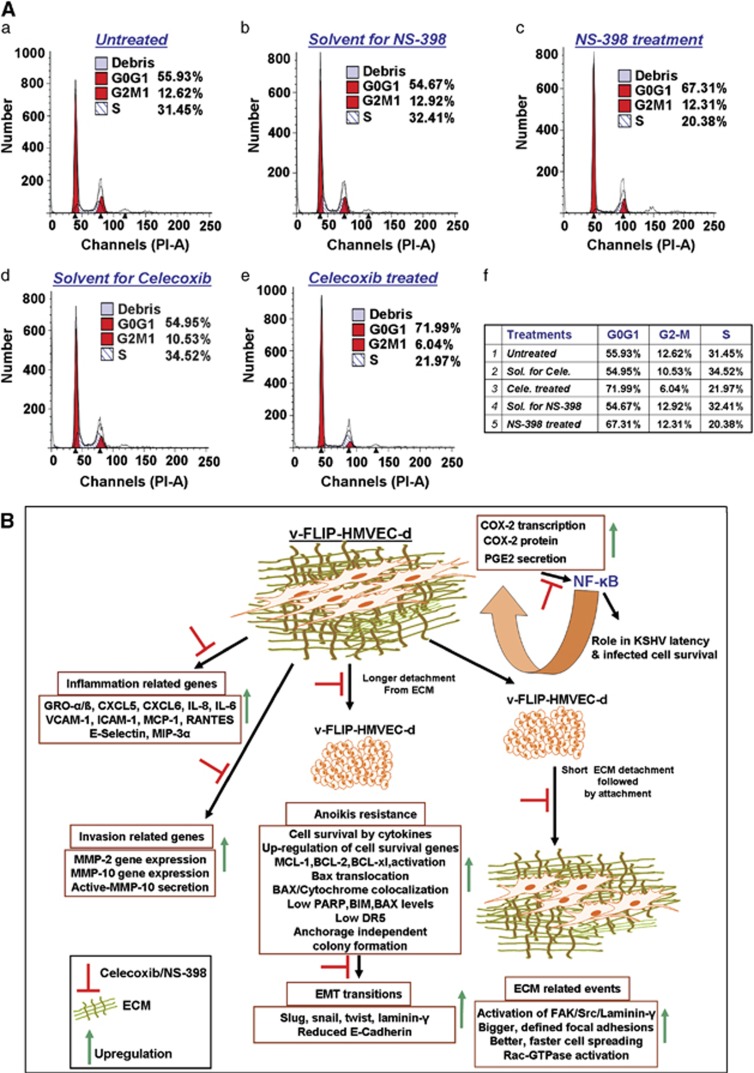
Kaposi's sarcoma herpesvirus (KSHV) latent oncoprotein viral FLICE (FADD-like interferon converting enzyme)-like inhibitory protein (v-FLIP) or K13, a potent activator of NF-κB, has well-established roles in KSHV latency and oncogenesis. KSHV-induced COX-2 represents a novel strategy employed by KSHV to promote latency and inflammation/angiogenesis/invasion. Here, we demonstrate that v-FLIP/K13 promotes tumorigenic effects via the induction of host protein COX-2 and its inflammatory metabolite PGE2 in an NF-κB-dependent manner. In addition to our previous studies demonstrating COX-2/PGE2's role in transcriptional regulation of KSHV latency promoter and latent gene expression, the current study adds to the complexity that though LANA-1 (latency associated nuclear antigen) is utilizing COX-2/PGE2 as critical factors for its transcriptional regulation, it is the v-FLIP/K13 gene in the KSHV latency cluster that maintains continuous COX-2/PGE2 levels in the infected cells. We demonstrate that COX-2 inhibition, via its chemical inhibitors (NS-398 or celecoxib), reduced v-FLIP/K13-mediated NF-κB induction, and extracellular matrix (ECM) interaction-mediated signaling, mitochondrial antioxidant enzyme manganese superoxide dismutase (MnSOD) levels, and subsequently downregulated detachment-induced apoptosis (anoikis) resistance. vFLIP expression mediated the secretion of cytokines, and spindle cell differentiation activated the phosphorylation of p38, RSK, FAK, Src, Akt and Rac1-GTPase. The COX-2 inhibition in v-FLIP/K13-HMVECs reduced inflammation and invasion/metastasis-related genes, along with reduced anchorage-independent colony formation via modulating 'extrinsic' as well as 'intrinsic' cell death pathways. COX-2 blockade in v-FLIP/K13-HMVEC cells drastically augmented cell death induced by removal of essential growth/survival factors secreted in the microenvironment. Transformed cells obtained from anchorage-independent colonies of COX-2 inhibitor-treated v-FLIP/K13-HMVEC cells expressed lower levels of endothelial-mesenchymal transition genes such as slug, snail and twist, and higher expression of the tumor-suppressor gene, E-cadherin. Taken together, our study provides strong evidences that FDA-approved COX-2 inhibitors have great potential in blocking tumorigenic events linked to KSHV's oncogenic protein v-FLIP/K13.
卡波西肉瘤疱疹病毒(KSHV)潜伏癌蛋白病毒 FLICE(FADD 样干扰素转换酶)样抑制蛋白(v-FLIP)或 K13,是 NF-κB 的一种有效激活剂,在 KSHV 潜伏和致癌作用中具有明确的作用。KSHV 诱导的 COX-2 代表了 KSHV 促进潜伏和炎症/血管生成/侵袭的一种新策略。在这里,我们证明 v-FLIP/K13 通过诱导宿主蛋白 COX-2 及其炎症代谢产物 PGE2 在 NF-κB 依赖性方式促进致瘤作用。除了我们之前的研究表明 COX-2/PGE2 在 KSHV 潜伏启动子和潜伏基因表达的转录调控中的作用外,本研究增加了复杂性,尽管 LANA-1(潜伏相关核抗原)正在利用 COX-2/PGE2 作为其转录调控的关键因素,但在 KSHV 潜伏群中,是 v-FLIP/K13 基因维持感染细胞中 COX-2/PGE2 的持续水平。我们证明,通过其化学抑制剂(NS-398 或塞来昔布)抑制 COX-2,可降低 v-FLIP/K13 介导的 NF-κB 诱导以及细胞外基质(ECM)相互作用介导的信号转导、线粒体抗氧化酶锰超氧化物歧化酶(MnSOD)水平,并随后下调脱落诱导的凋亡(失巢凋亡)抵抗。vFLIP 表达介导细胞因子的分泌,纺锤体细胞分化激活 p38、RSK、FAK、Src、Akt 和 Rac1-GTPase 的磷酸化。COX-2 抑制在 v-FLIP/K13-HMVEC 中降低了炎症和侵袭/转移相关基因的表达,并通过调节“外在”和“内在”细胞死亡途径减少了锚定独立集落的形成。COX-2 抑制剂在 v-FLIP/K13-HMVEC 细胞中的阻断大大增加了去除微环境中分泌的必需生长/存活因子诱导的细胞死亡。从 COX-2 抑制剂处理的 v-FLIP/K13-HMVEC 细胞的锚定独立集落获得的转化细胞表达较低水平的内皮-间充质转化基因,如 slug、snail 和 twist,以及较高水平的肿瘤抑制基因 E-cadherin。总之,我们的研究提供了强有力的证据表明,美国食品和药物管理局批准的 COX-2 抑制剂在阻断与 KSHV 致癌蛋白 v-FLIP/K13 相关的致癌事件方面具有巨大潜力。