Department of Pharmacology and Systems Therapeutics, Icahn School of Medicine at Mount Sinai, One Gustave L, Levy Place, Box 1215, New York, NY 10029, USA.
BMC Bioinformatics. 2013 Apr 15;14:128. doi: 10.1186/1471-2105-14-128.
System-wide profiling of genes and proteins in mammalian cells produce lists of differentially expressed genes/proteins that need to be further analyzed for their collective functions in order to extract new knowledge. Once unbiased lists of genes or proteins are generated from such experiments, these lists are used as input for computing enrichment with existing lists created from prior knowledge organized into gene-set libraries. While many enrichment analysis tools and gene-set libraries databases have been developed, there is still room for improvement.
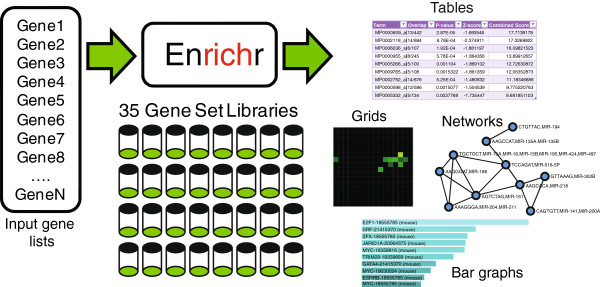
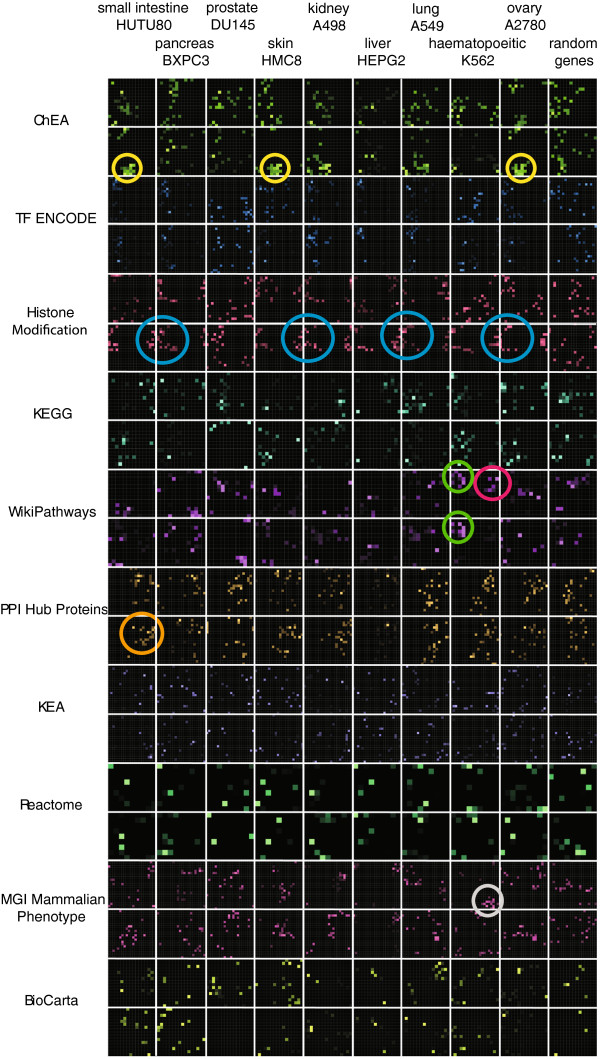
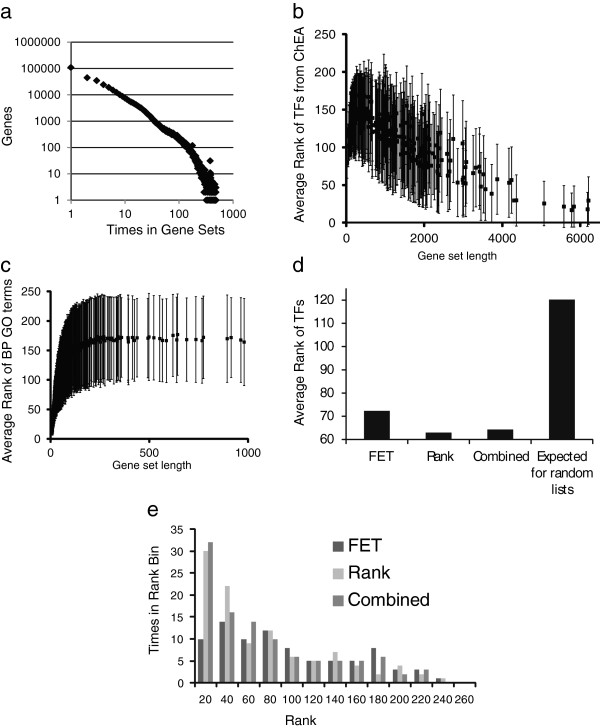
Here, we present Enrichr, an integrative web-based and mobile software application that includes new gene-set libraries, an alternative approach to rank enriched terms, and various interactive visualization approaches to display enrichment results using the JavaScript library, Data Driven Documents (D3). The software can also be embedded into any tool that performs gene list analysis. We applied Enrichr to analyze nine cancer cell lines by comparing their enrichment signatures to the enrichment signatures of matched normal tissues. We observed a common pattern of up regulation of the polycomb group PRC2 and enrichment for the histone mark H3K27me3 in many cancer cell lines, as well as alterations in Toll-like receptor and interlukin signaling in K562 cells when compared with normal myeloid CD33+ cells. Such analyses provide global visualization of critical differences between normal tissues and cancer cell lines but can be applied to many other scenarios.
Enrichr is an easy to use intuitive enrichment analysis web-based tool providing various types of visualization summaries of collective functions of gene lists. Enrichr is open source and freely available online at: http://amp.pharm.mssm.edu/Enrichr.
对哺乳动物细胞中的基因和蛋白质进行系统水平的分析会产生差异表达基因/蛋白质的列表,需要进一步分析它们的集体功能,以提取新知识。一旦从这些实验中生成了无偏差的基因或蛋白质列表,就可以将这些列表用作计算与现有基因集库中基于先验知识创建的列表进行富集的输入。虽然已经开发了许多富集分析工具和基因集库数据库,但仍有改进的空间。
在这里,我们介绍了 Enrichr,这是一个集成的基于网络和移动的软件应用程序,它包含了新的基因集库、一种对丰富术语进行排名的替代方法,以及各种交互式可视化方法,使用 JavaScript 库 Data Driven Documents (D3) 来显示富集结果。该软件还可以嵌入到执行基因列表分析的任何工具中。我们应用 Enrichr 通过将九个癌细胞系的富集特征与匹配的正常组织的富集特征进行比较来进行分析。我们观察到许多癌细胞系中多梳组 PRC2 的上调和组蛋白标记 H3K27me3 的富集的共同模式,以及与正常髓样 CD33+细胞相比 K562 细胞中 Toll 样受体和白细胞介素信号的改变。这种分析提供了正常组织和癌细胞系之间关键差异的全局可视化,但可以应用于许多其他情况。
Enrichr 是一种易于使用的直观富集分析网络工具,提供了基因列表集体功能的各种类型的可视化摘要。Enrichr 是开源的,可免费在线获取:http://amp.pharm.mssm.edu/Enrichr。