Centre for GeoGenetics, Natural History Museum of Denmark, University of Copenhagen, 1350 København K, Denmark.
Bioinformatics. 2013 Jul 1;29(13):1682-4. doi: 10.1093/bioinformatics/btt193. Epub 2013 Apr 23.
Ancient DNA (aDNA) molecules in fossilized bones and teeth, coprolites, sediments, mummified specimens and museum collections represent fantastic sources of information for evolutionary biologists, revealing the agents of past epidemics and the dynamics of past populations. However, the analysis of aDNA generally faces two major issues. Firstly, sequences consist of a mixture of endogenous and various exogenous backgrounds, mostly microbial. Secondly, high nucleotide misincorporation rates can be observed as a result of severe post-mortem DNA damage. Such misincorporation patterns are instrumental to authenticate ancient sequences versus modern contaminants. We recently developed the user-friendly mapDamage package that identifies such patterns from next-generation sequencing (NGS) sequence datasets. The absence of formal statistical modeling of the DNA damage process, however, precluded rigorous quantitative comparisons across samples.
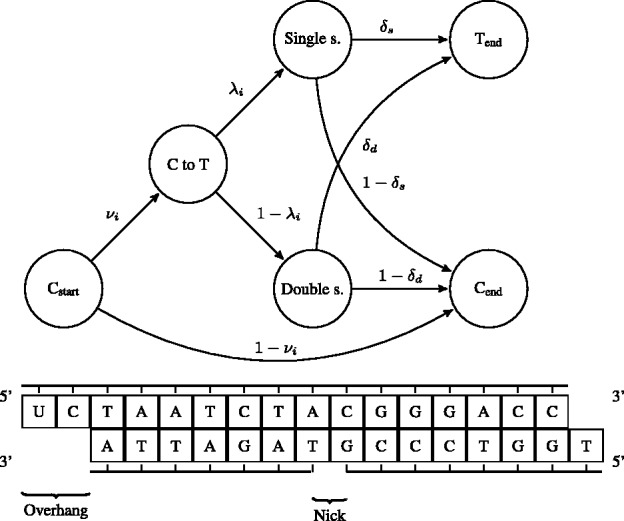
Here, we describe mapDamage 2.0 that extends the original features of mapDamage by incorporating a statistical model of DNA damage. Assuming that damage events depend only on sequencing position and post-mortem deamination, our Bayesian statistical framework provides estimates of four key features of aDNA molecules: the average length of overhangs (λ), nick frequency (ν) and cytosine deamination rates in both double-stranded regions ( ) and overhangs ( ). Our model enables rescaling base quality scores according to their probability of being damaged. mapDamage 2.0 handles NGS datasets with ease and is compatible with a wide range of DNA library protocols.
mapDamage 2.0 is available at ginolhac.github.io/mapDamage/ as a Python package and documentation is maintained at the Centre for GeoGenetics Web site (geogenetics.ku.dk/publications/mapdamage2.0/).
Supplementary data are available at Bioinformatics online.
来自化石骨骼和牙齿、粪化石、沉积物、木乃伊标本和博物馆藏品中的古代 DNA(aDNA)分子为进化生物学家提供了极好的信息来源,揭示了过去传染病的病原体和过去人口的动态。然而,aDNA 的分析通常面临两个主要问题。首先,序列由内源性和各种外源性背景(主要是微生物)的混合物组成。其次,由于严重的死后 DNA 损伤,可能会观察到高核苷酸错配率。这些错配模式对于验证古代序列与现代污染物非常重要。我们最近开发了用户友好的 mapDamage 包,可从下一代测序(NGS)序列数据集中识别这些模式。然而,由于缺乏对 DNA 损伤过程的正式统计建模,因此无法在样本之间进行严格的定量比较。
在这里,我们描述了 mapDamage 2.0,它通过纳入 DNA 损伤的统计模型扩展了 mapDamage 的原始功能。假设损伤事件仅取决于测序位置和死后脱氨作用,我们的贝叶斯统计框架提供了四个关键特征的估计值:双链区域( )和突出端( )中 aDNA 分子的平均突出端长度(λ)、缺口频率(ν)和胞嘧啶脱氨率。我们的模型可以根据其受损的概率重新调整碱基质量得分。mapDamage 2.0 易于处理 NGS 数据集,并且与广泛的 DNA 文库协议兼容。
mapDamage 2.0 可作为 Python 包在 ginolhac.github.io/mapDamage/上获得,文档在哥本哈根大学遗传中心网站(geogenetics.ku.dk/publications/mapdamage2.0/)上维护。
补充数据可在 Bioinformatics 在线获得。