Department of Medical Microbiology, Maastricht University Medical Centre+ Maastricht, Netherlands.
Front Microbiol. 2013 Apr 17;4:87. doi: 10.3389/fmicb.2013.00087. eCollection 2013.
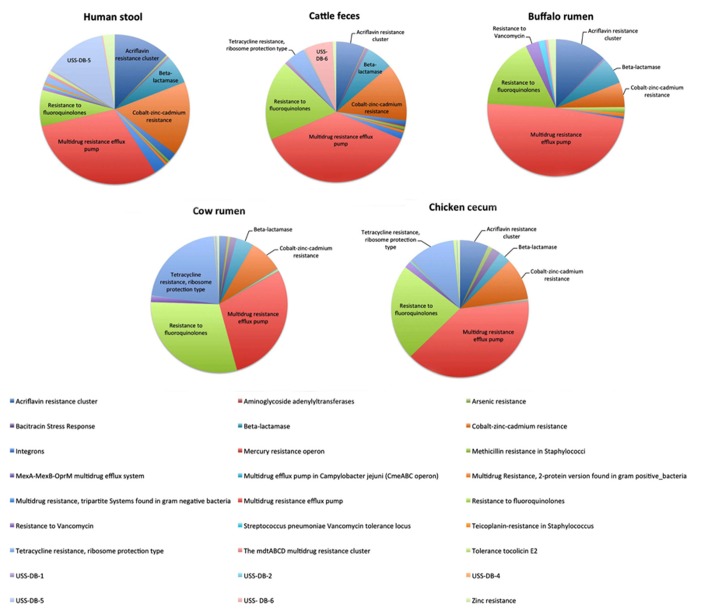
The gut microbiota is amongst the most densely populated microbial ecosystem on earth. While the microbiome exerts numerous health beneficial functions, the high density of micro-organisms within this ecosystem also facilitates horizontal transfer of antimicrobial resistance (AMR) genes to potential pathogenic bacteria. Over the past decades antibiotic susceptibility testing of specific indicator bacteria from the microbiome, such as Escherichia coli, has been the method of choice in most studies. These studies have greatly enlarged our understanding on the prevalence and distribution of AMR and associated risk factors. Recent studies using (functional) metagenomics, however, highlighted the unappreciated diversity of AMR genes in the human microbiome and identified genes that had not been described previously. Next to metagenomics, more targeted approaches such as polymerase chain reaction for detection and quantification of AMR genes within a population are promising, in particular for large-scale epidemiological screening. Here we present an overview of the indigenous microbiota as a reservoir of AMR genes, the current knowledge on this "resistome" and the recent and upcoming advances in the molecular diagnostic approaches to unravel this reservoir.
肠道微生物群是地球上微生物密度最高的生态系统之一。虽然微生物组发挥了许多有益健康的功能,但这个生态系统中微生物的高密度也促进了抗生素耐药性 (AMR) 基因向潜在病原菌的水平转移。在过去的几十年中,对微生物组中特定指示菌(如大肠杆菌)的抗生素药敏试验一直是大多数研究的首选方法。这些研究极大地提高了我们对 AMR 的流行和分布以及相关危险因素的认识。然而,最近使用(功能)宏基因组学的研究强调了人类微生物组中 AMR 基因的多样性尚未被充分认识,并确定了以前未描述过的基因。除了宏基因组学之外,更具针对性的方法,如聚合酶链反应检测和定量人群中的 AMR 基因,在大规模流行病学筛查中具有很大的应用前景。本文概述了 AMR 基因的本土微生物群作为其储库,以及目前对这个“耐药组”的认识,以及在揭示这个储库方面的分子诊断方法的最新进展。