Centre for Infectious Diseases and Microbiology and Sydney Institute for Emerging Infections and Biosecurity, University of Sydney, Westmead Hospital, Hawkesbury Road, Westmead, NSW 2145, Australia.
BMC Bioinformatics. 2013 May 1;14:148. doi: 10.1186/1471-2105-14-148.
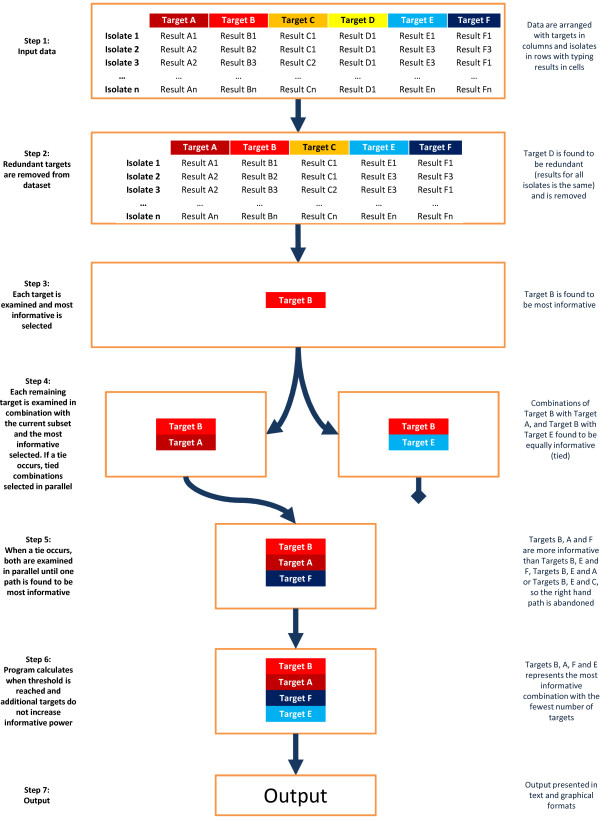
High-throughput sequencing can identify numerous potential genomic targets for microbial strain typing, but identification of the most informative combinations requires the use of computational screening tools. This paper describes novel software-- Automated Selection of Typing Target Subsets (AuSeTTS)--that allows intelligent selection of optimal targets for pathogen strain typing. The objective of this software is to maximise both discriminatory power, using Simpson's index of diversity (D), and concordance with existing typing methods, using the adjusted Wallace coefficient (AW). The program interrogates molecular typing results for panels of isolates, based on large target sets, and iteratively examines each target, one-by-one, to determine the most informative subset.
AuSeTTS was evaluated using three target sets: 51 binary targets (13 toxin genes, 16 phage-related loci and 22 SCCmec elements), used for multilocus typing of 153 methicillin-resistant Staphylococcus aureus (MRSA) isolates; 17 MLVA loci in 502 Streptococcus pneumoniae isolates from the MLVA database (http://www.mlva.eu) and 12 MLST loci for 98 Cryptococcus spp. isolates.The maximum D for MRSA, 0.984, was achieved with a subset of 20 targets and a D value of 0.954 with 7 targets. Twelve targets predicted MLST with a maximum AW of 0.9994. All 17 S. pneumoniae MLVA targets were required to achieve maximum D of 0.997, but 4 targets reached D of 0.990. Twelve targets predicted pneumococcal serotype with a maximum AW of 0.899 and 9 predicted MLST with maximum AW of 0.963. Eight of the 12 MLST loci were sufficient to achieve the maximum D of 0.963 for Cryptococcus spp.
Computerised analysis with AuSeTTS allows rapid selection of the most discriminatory targets for incorporation into typing schemes. Output of the program is presented in both tabular and graphical formats and the software is available for free download from http://www.cidmpublichealth.org/pages/ausetts.html.
高通量测序可以识别大量潜在的微生物菌株分型的基因组靶标,但识别最具信息量的组合需要使用计算筛选工具。本文描述了一种新的软件——自动选择分型靶标子集(AuSeTTS),该软件允许对病原体菌株分型的最佳目标进行智能选择。该软件的目标是最大化多样性指数(D)的区分能力,使用 Simpson 指数,以及与现有分型方法的一致性,使用调整 Wallace 系数(AW)。该程序根据大型靶标集,对分离株的分子分型结果进行查询,并迭代地逐个检查每个靶标,以确定最具信息量的子集。
使用三个靶标集评估了 AuSeTTS:51 个二进制靶标(13 个毒素基因、16 个噬菌体相关基因座和 22 个 SCCmec 元件),用于对 153 株耐甲氧西林金黄色葡萄球菌(MRSA)分离株进行多位点分型;502 株肺炎链球菌分离株的 17 个 MLVA 基因座来自 MLVA 数据库(http://www.mlva.eu)和 98 株隐球菌的 12 个 MLST 基因座。MRSA 的最大 D 值为 0.984,使用 20 个目标子集实现,D 值为 0.954,使用 7 个目标。12 个目标预测 MLST,最大 AW 值为 0.9994。要实现 0.997 的最大 D 值,需要所有 17 个 S. pneumoniae MLVA 靶标,但 4 个靶标达到了 0.990 的 D 值。12 个目标预测肺炎链球菌血清型,最大 AW 值为 0.899,12 个预测 MLST,最大 AW 值为 0.963。12 个 MLST 基因座中的 8 个足以达到隐球菌的最大 D 值 0.963。
使用 AuSeTTS 进行计算机分析可以快速选择最具区分能力的靶标纳入分型方案。程序的输出以表格和图形格式呈现,该软件可从 http://www.cidmpublichealth.org/pages/ausetts.html 免费下载。