BMC Genomics. 2013 May 7;14:306. doi: 10.1186/1471-2164-14-306.
About 80% of today's land plants are able to establish an arbuscular mycorrhizal (AM) symbiosis with Glomeromycota fungi to improve their access to nutrients and water in the soil. On the molecular level, the development of AM symbioses is only partly understood, due to the asynchronous development of the microsymbionts in the host roots. Although many genes specifically activated during fungal colonization have been identified, genome-wide information on the exact place and time point of their activation remains limited.
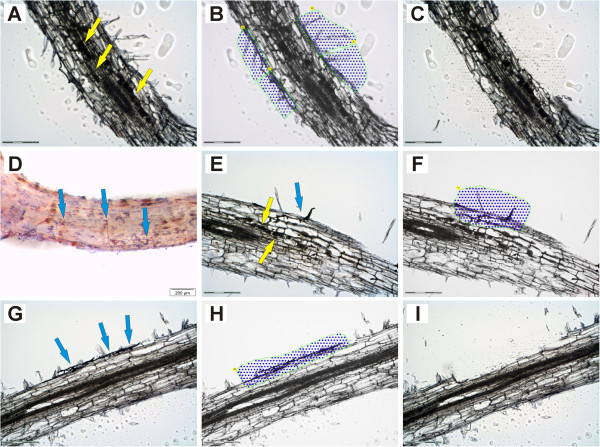
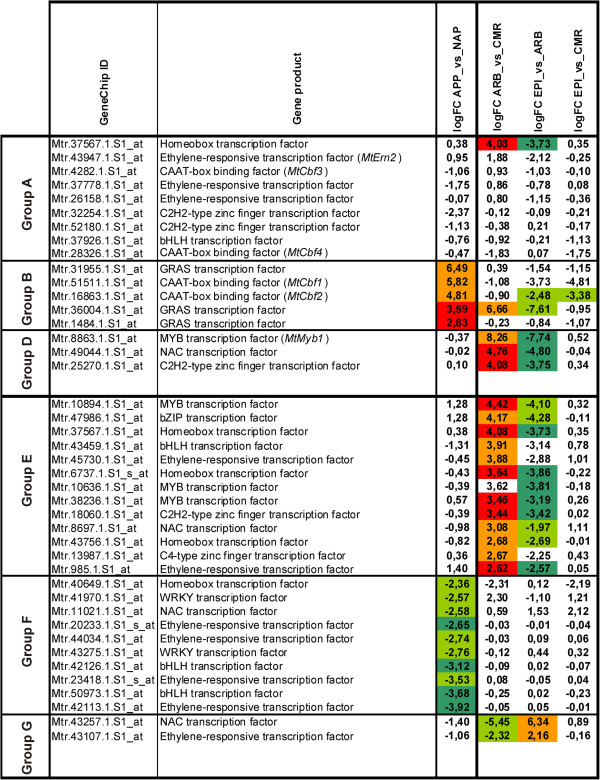
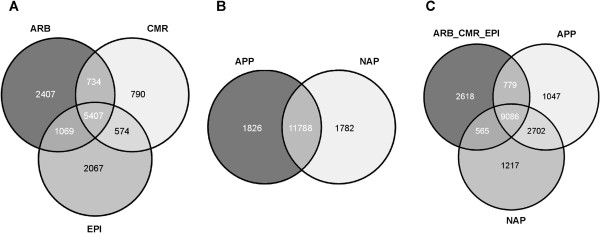
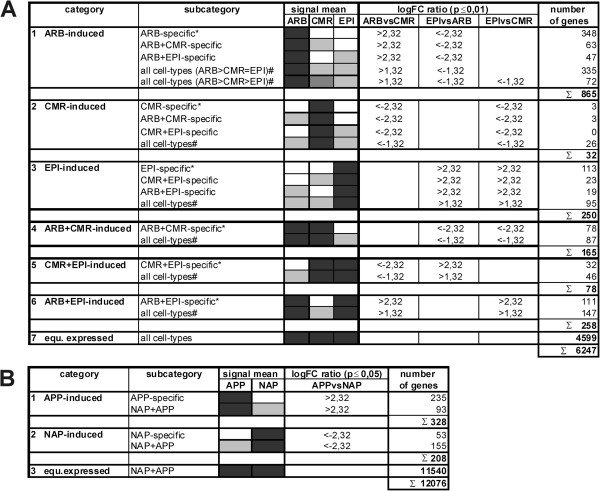
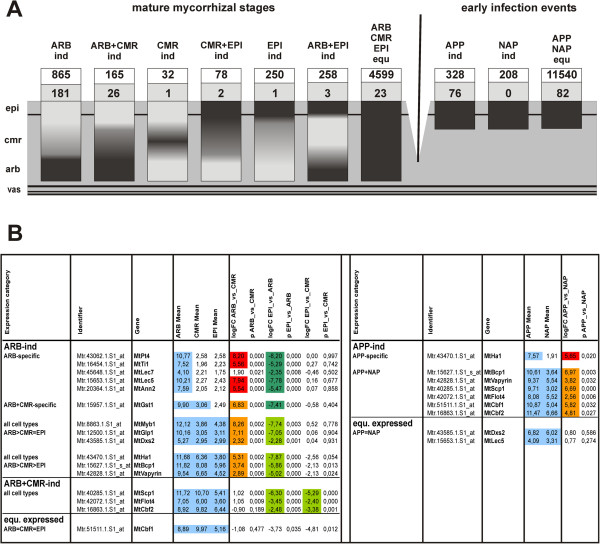
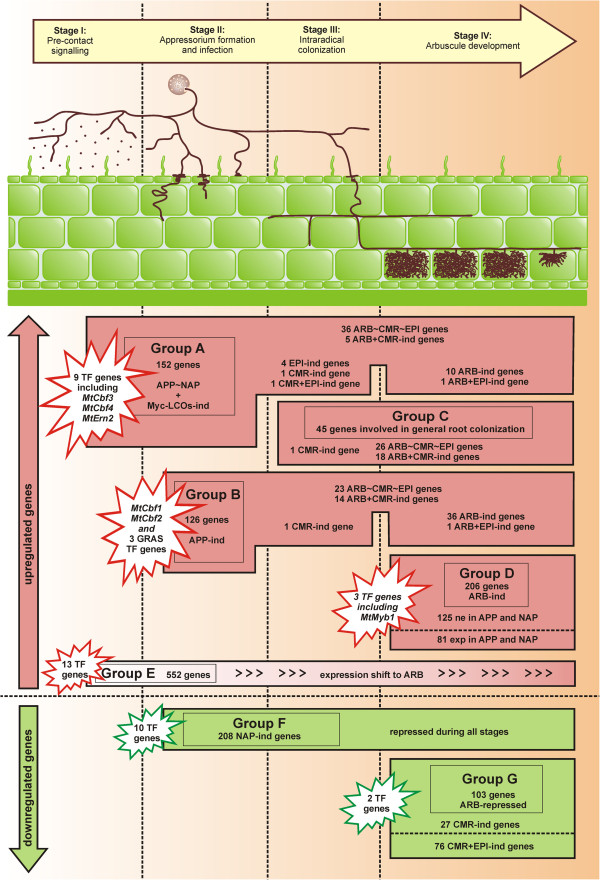
In this study, we relied on a combination of laser-microdissection and the use of Medicago GeneChips to perform a genome-wide analysis of transcription patterns in defined cell-types of Medicago truncatula roots mycorrhized with Glomus intraradices. To cover major stages of AM development, we harvested cells at 5-6 and at 21 days post inoculation (dpi). Early developmental stages of the AM symbiosis were analysed by monitoring gene expression in appressorial and non-appressorial areas from roots harbouring infection units at 5-6 dpi. Here, the use of laser-microdissection for the first time enabled the targeted harvest of those sites, where fungal hyphae first penetrate the root. Circumventing contamination with developing arbuscules, we were able to specifically detect gene expression related to early infection events. To cover the late stages of AM formation, we studied arbusculated cells, cortical cells colonized by intraradical hyphae, and epidermal cells from mature mycorrhizal roots at 21 dpi. Taken together, the cell-specific expression patterns of 18014 genes were revealed, including 1392 genes whose transcription was influenced by mycorrhizal colonization at different stages, namely the pre-contact phase, the infection of roots via fungal appressoria, the subsequent colonization of the cortex by fungal hyphae, and finally the formation of arbuscules. Our cellular expression patterns identified distinct groups of AM-activated genes governing the sequential reprogramming of host roots towards an accommodation of microsymbionts, including 42 AM-activated transcription factor genes.
Our genome-wide analysis provides novel information on the cell-specific activity of AM-activated genes during both early and late stages of AM development, together revealing the road map of fine-tuned adjustments of transcript accumulation within root tissues during AM fungal colonization.
今天约 80%的陆地植物能够与 Glomeromycota 真菌建立丛枝菌根(AM)共生关系,以改善其对土壤中养分和水分的获取。 在分子水平上,由于宿主根系中小微生物的发育不同步,AM 共生的发育仅部分被理解。 虽然已经鉴定出许多在真菌定殖过程中特异性激活的基因,但关于其激活的确切位置和时间点的全基因组信息仍然有限。
在这项研究中,我们依赖于激光显微切割和 Medicago GeneChips 的使用,对与 Glomus intraradices 共生的 Medicago truncatula 根的特定细胞类型中的转录模式进行了全基因组分析。 为了涵盖 AM 发育的主要阶段,我们在接种后 5-6 天和 21 天收获细胞(dpi)。 通过监测 5-6dpi 时含有感染单位的根中附着器和非附着器区域的基因表达,分析 AM 共生的早期发育阶段。 在这里,激光显微切割的首次使用使我们能够有针对性地收获真菌菌丝首次穿透根的那些部位。 避免与发育中的丛枝共染,我们能够特异性地检测与早期感染事件相关的基因表达。 为了涵盖 AM 形成的后期阶段,我们研究了丛枝细胞、根内真菌菌丝定殖的皮层细胞和成熟共生根的表皮细胞在 21dpi 时。 总共揭示了 18014 个基因的细胞特异性表达模式,包括 1392 个转录受不同阶段菌根定殖影响的基因,即接触前阶段、真菌附着器感染根、随后真菌菌丝定殖皮层和最后丛枝的形成。 我们的细胞表达模式确定了 AM 激活基因的不同亚群,这些基因控制着宿主根向适应微生物的顺序重编程,包括 42 个 AM 激活转录因子基因。
我们的全基因组分析提供了 AM 激活基因在 AM 发育的早期和晚期阶段的细胞特异性活性的新信息,共同揭示了在 AM 真菌定殖过程中,根组织中转录物积累的精细调整路线图。