Department of Immunology, Genetics and Pathology, Rudbeck Laboratory, Uppsala University, Uppsala SE-751 85, Sweden.
BMC Cancer. 2013 May 9;13:231. doi: 10.1186/1471-2407-13-231.
Aggressive neuroblastoma remains a significant cause of childhood cancer death despite current intensive multimodal treatment protocols. The purpose of the present work was to characterize the genetic and clinical diversity of such tumors by high resolution arrayCGH profiling.
Based on a 32K BAC whole-genome tiling path array and using 50-250K Affymetrix SNP array platforms for verification, DNA copy number profiles were generated for 34 consecutive high-risk or lethal outcome neuroblastomas. In addition, age and MYCN amplification (MNA) status were retrieved for 112 unfavorable neuroblastomas of the Swedish Childhood Cancer Registry, representing a 25-year neuroblastoma cohort of Sweden, here used for validation of the findings. Statistical tests used were: Fisher's exact test, Bayes moderated t-test, independent samples t-test, and correlation analysis.
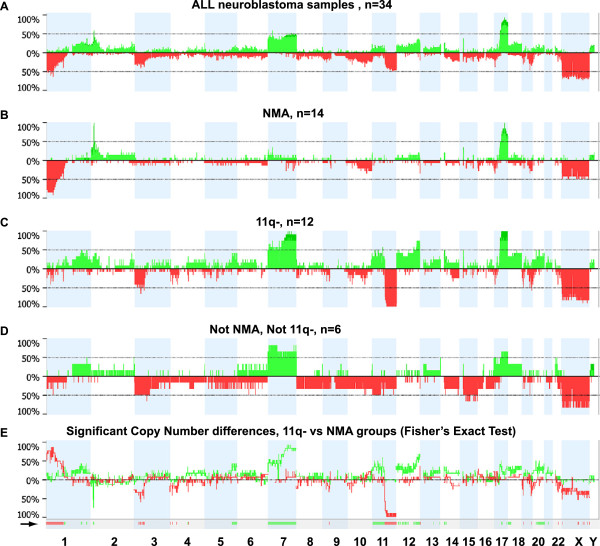
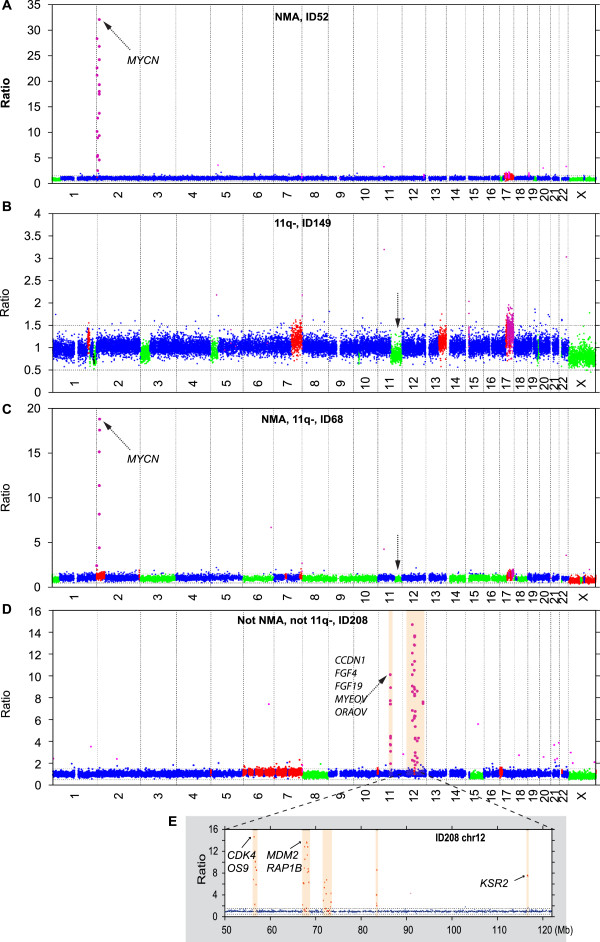
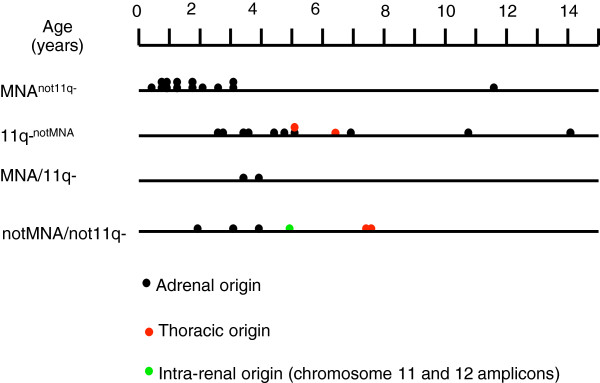
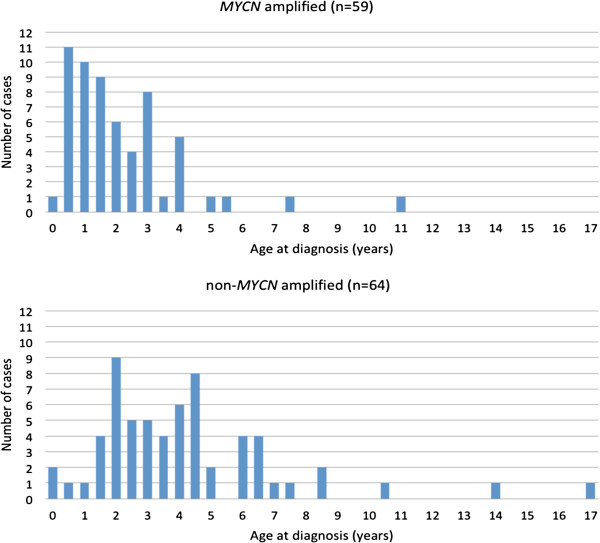
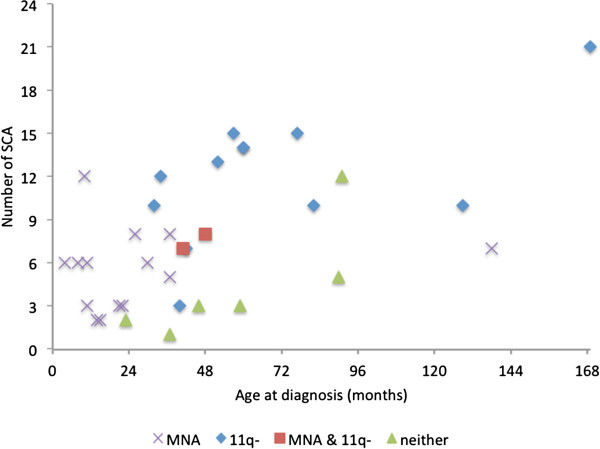
MNA or segmental 11q loss (11q-) was found in 28/34 tumors. With two exceptions, these aberrations were mutually exclusive. Children with MNA tumors were diagnosed at significantly younger ages than those with 11q- tumors (mean: 27.4 vs. 69.5 months; p=0.008; n=14/12), and MNA tumors had significantly fewer segmental chromosomal aberrations (mean: 5.5 vs. 12.0; p<0.001). Furthermore, in the 11q- tumor group a positive correlation was seen between the number of segmental aberrations and the age at diagnosis (Pearson Correlation 0.606; p=0.037). Among nonMNA/non11q- tumors (n=6), one tumor displayed amplicons on 11q and 12q and three others bore evidence of progression from low-risk tumors due to retrospective evidence of disease six years before diagnosis, or due to tumor profiles with high proportions of numerical chromosomal aberrations. An early age at diagnosis of MNA neuroblastomas was verified by registry data, with an average of 29.2 months for 43 cases that were not included in the present study.
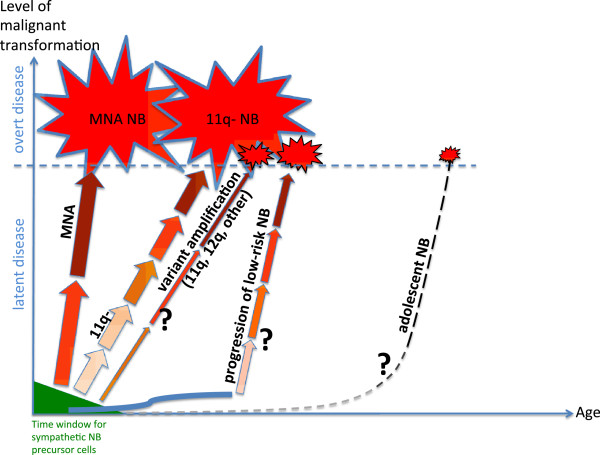
MNA and segmental 11q loss define two major genetic variants of unfavorable neuroblastoma with apparent differences in their pace of tumor evolution and in genomic integrity. Other possible, but less common, routes in the development of aggressive tumors are progression of low-risk infant-type lesions, and gene amplifications other than MYCN. Knowledge on such nosological diversity of aggressive neuroblastoma might influence future strategies for therapy.
尽管目前采用了强化的多模式治疗方案,侵袭性神经母细胞瘤仍然是儿童癌症死亡的一个重要原因。本研究的目的是通过高分辨率的阵列 CGH 分析来描述此类肿瘤的遗传和临床多样性。
基于 32K BAC 全基因组平铺路径阵列,并使用 50-250K Affymetrix SNP 阵列平台进行验证,为 34 例连续的高危或致死性神经母细胞瘤生成了 DNA 拷贝数谱。此外,还为瑞典儿童癌症登记处的 112 例不利神经母细胞瘤检索了年龄和 MYCN 扩增(MNA)状态,这些肿瘤代表了瑞典 25 年的神经母细胞瘤队列,在此用于验证研究结果。使用的统计检验包括:Fisher 确切检验、贝叶斯校正 t 检验、独立样本 t 检验和相关分析。
在 34 例肿瘤中发现了 MNA 或片段性 11q 缺失(11q-)。除了两个例外,这些异常是相互排斥的。MNA 肿瘤患儿的诊断年龄明显小于 11q-肿瘤患儿(平均年龄:27.4 与 69.5 个月;p=0.008;n=14/12),且 MNA 肿瘤的片段性染色体异常明显较少(平均数量:5.5 与 12.0;p<0.001)。此外,在 11q-肿瘤组中,诊断时的片段性异常数量与年龄之间存在正相关(Pearson 相关系数 0.606;p=0.037)。在非 MNA/非 11q-肿瘤(n=6)中,有 1 例肿瘤显示 11q 和 12q 的扩增子,另外 3 例由于诊断前六年的疾病回顾性证据或由于肿瘤谱中存在高比例的数值染色体异常,表现出从低危肿瘤进展的证据。通过登记数据验证了 MNA 神经母细胞瘤的早诊年龄,43 例未纳入本研究的病例平均为 29.2 个月。
MNA 和片段性 11q 缺失定义了不良神经母细胞瘤的两个主要遗传变异体,它们在肿瘤进化速度和基因组完整性方面存在明显差异。其他可能的、但不太常见的侵袭性肿瘤发展途径是低危婴儿型病变的进展,以及除 MYCN 以外的基因扩增。了解侵袭性神经母细胞瘤的这种分类学多样性可能会影响未来的治疗策略。