Department of Physiology, The University of Tennessee Health Science Center, Memphis, Tennessee, United States of America.
PLoS One. 2013 May 10;8(5):e63465. doi: 10.1371/journal.pone.0063465. Print 2013.
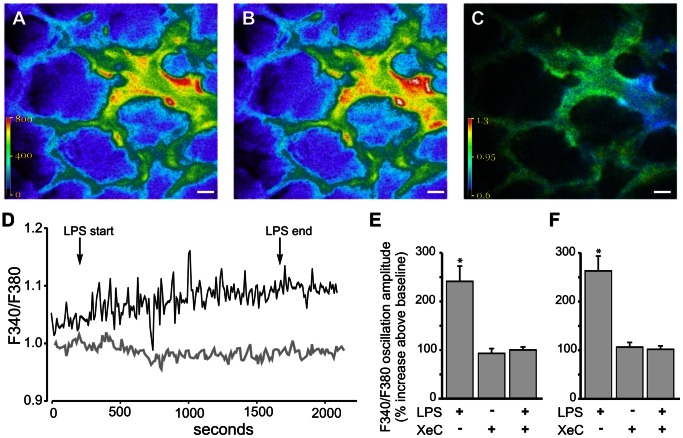
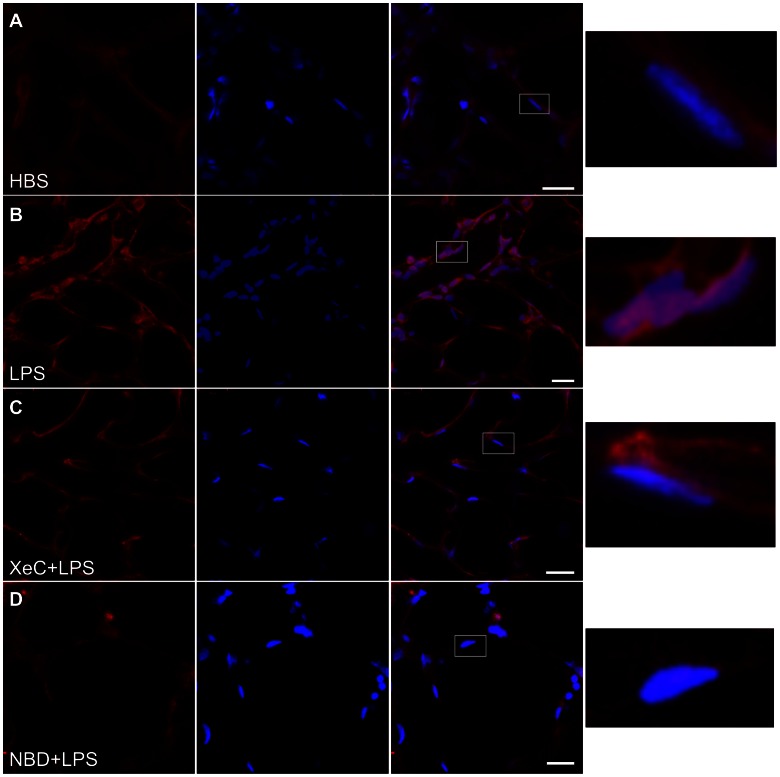
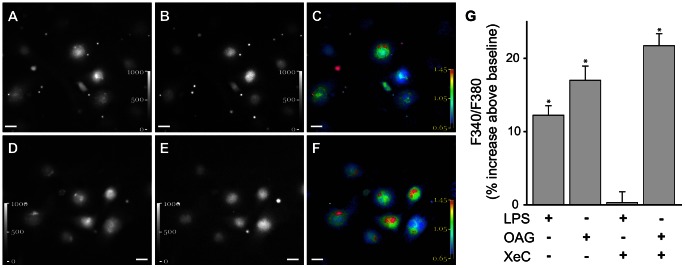
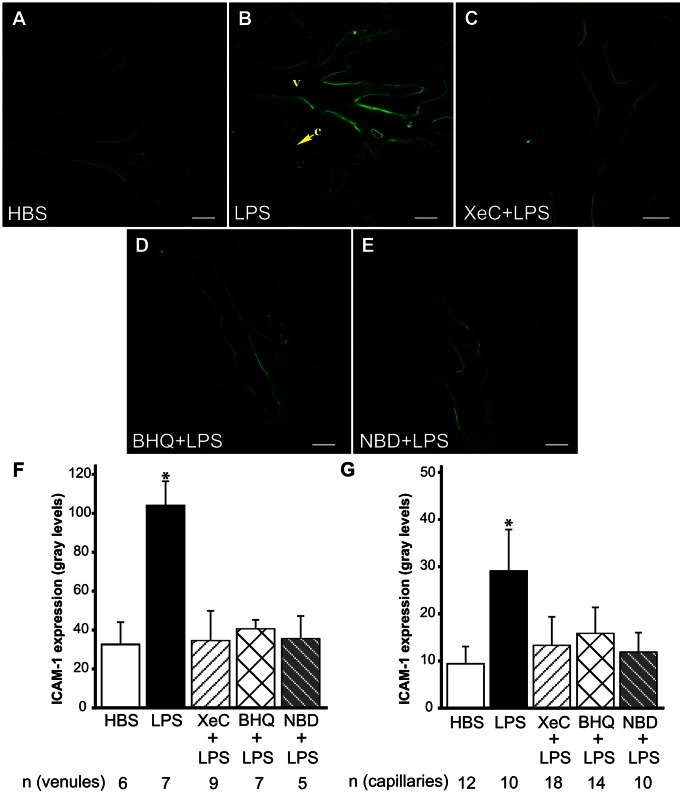
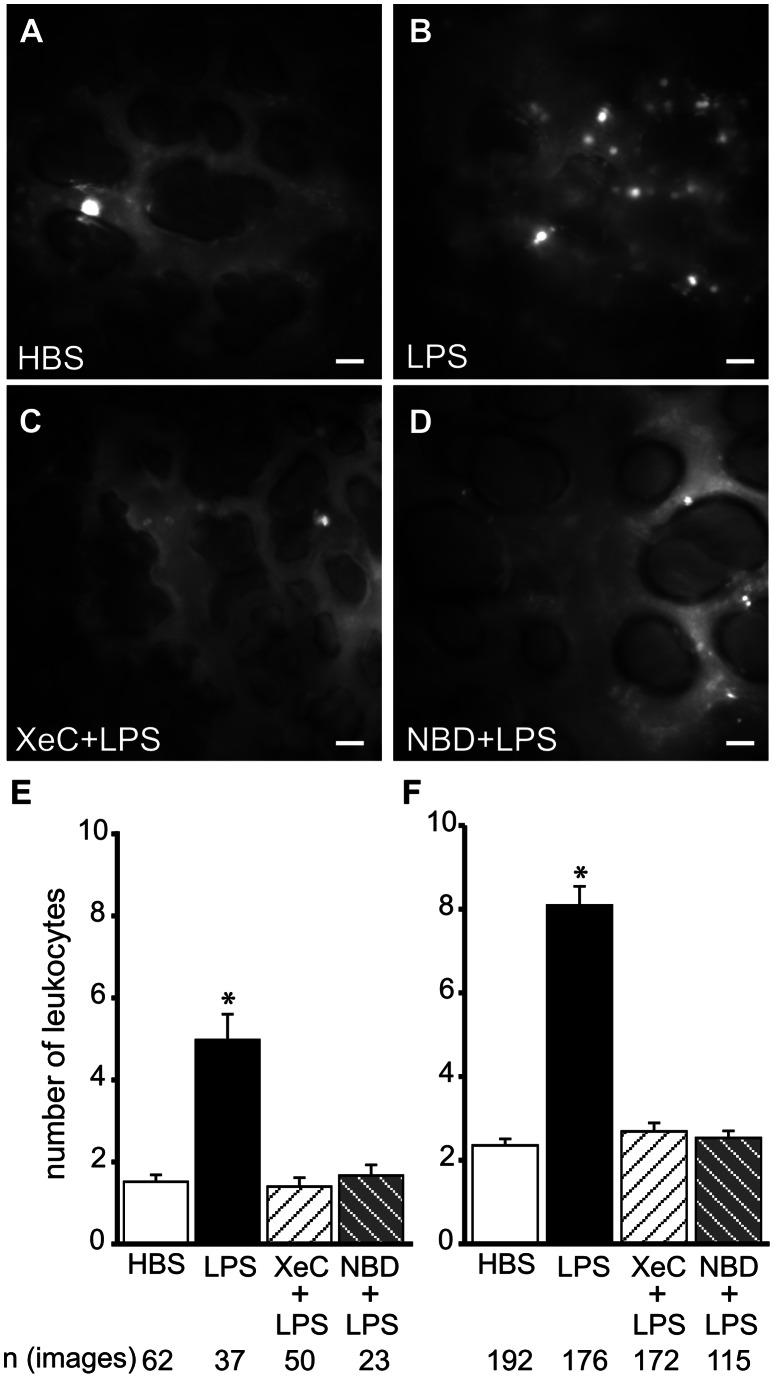
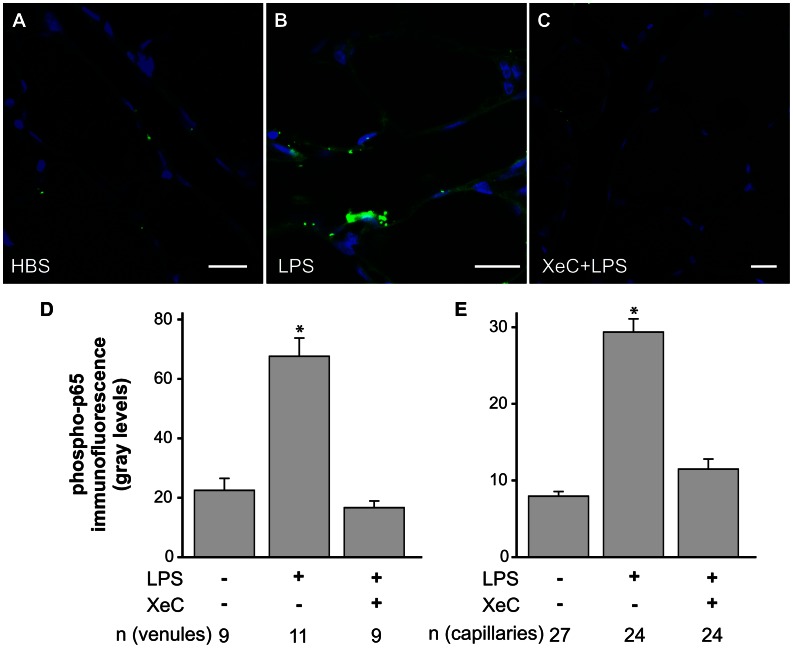
The pulmonary microvasculature plays a critical role in endotoxin-induced acute lung injury. However, the relevant signaling remain unclear. Specifically the role of endothelial Ca(2+) in the induction of endotoxin-mediated responses in lung microvessels remains undefined. Toward elucidating this, we used the isolated blood-perfused rat lung preparation. We loaded microvessels with the Ca(2+) indicator, Fura 2 AM and then determined Ca(2+) responses to infusions of lipopolysaccharide (LPS) into the microvessels. LPS induced a more than two-fold increase in the amplitude of cytosolic Ca(2+) oscillations. Inhibiting inositol 1,4,5 trisphosphate receptors on endoplasmic reticulum (ER) Ca(2+) stores with Xestospongin C (XeC), blocked the LPS-induced increase in the Ca(2+) oscillation amplitude. However, XeC did not affect entry of external Ca(2+) via plasma membrane Ca(2+) channels in lung microvascular endothelial cells. This suggested that LPS augmented the oscillations via release of Ca(2+) from ER stores. In addition, XeC also blocked LPS-mediated activation and nuclear translocation of nuclear factor-kappa B in lung microvessels. Further, inhibiting ER Ca(2+) release blunted increases in intercellular adhesion molecule-1 expression and retention of naïve leukocytes in LPS-treated microvessels. Taken together, the data suggest that LPS-mediated Ca(2+) release from ER stores underlies nuclear factor-kappa B activation and downstream inflammatory signaling in lung microvessels. Thus, we show for the first time a role for inositol 1,4,5 trisphosphate-mediated ER Ca(2+) release in the induction of LPS responses in pulmonary microvascular endothelium. Mechanisms that blunt this signaling may mitigate endotoxin-induced morbidity.
肺微血管在脂多糖(内毒素)诱导的急性肺损伤中起着关键作用。然而,相关信号通路仍不清楚。具体来说,内皮细胞 Ca(2+) 在诱导肺微血管中内毒素介导的反应中的作用仍未确定。为了阐明这一点,我们使用了分离的血液灌注大鼠肺制备物。我们将 Ca(2+)指示剂 Fura 2 AM 加载到微血管中,然后确定向微血管中注入脂多糖(LPS)时 Ca(2+)的反应。LPS 诱导细胞浆 Ca(2+) 振荡幅度增加两倍以上。用 Xestospongin C(XeC)抑制内质网(ER)Ca(2+) 库中的肌醇 1,4,5 三磷酸受体,阻断了 LPS 诱导的 Ca(2+) 振荡幅度增加。然而,XeC 不影响肺微血管内皮细胞质膜 Ca(2+) 通道的外 Ca(2+) 内流。这表明 LPS 通过从 ER 库中释放 Ca(2+) 来增强振荡。此外,XeC 还阻断了 LPS 介导的肺微血管中核因子-κB 的激活和核转位。此外,抑制 ER Ca(2+) 释放可减弱 LPS 处理的微血管中细胞间黏附分子-1 的表达增加和幼稚白细胞的保留。综上所述,数据表明 LPS 介导的 ER 库中 Ca(2+) 的释放是核因子-κB 在肺微血管内皮细胞中激活和下游炎症信号的基础。因此,我们首次表明肌醇 1,4,5 三磷酸介导的 ER Ca(2+) 释放在内毒素诱导的肺微血管内皮细胞 LPS 反应的诱导中起作用。减轻这种信号的机制可能会减轻内毒素引起的发病率。