INRIA Sierra Project-Team, Paris F-75013, France.
BMC Bioinformatics. 2013 May 22;14:164. doi: 10.1186/1471-2105-14-164.
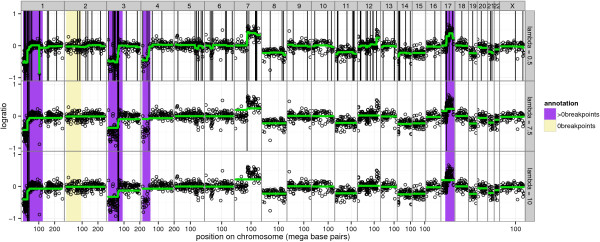
Many models have been proposed to detect copy number alterations in chromosomal copy number profiles, but it is usually not obvious to decide which is most effective for a given data set. Furthermore, most methods have a smoothing parameter that determines the number of breakpoints and must be chosen using various heuristics.
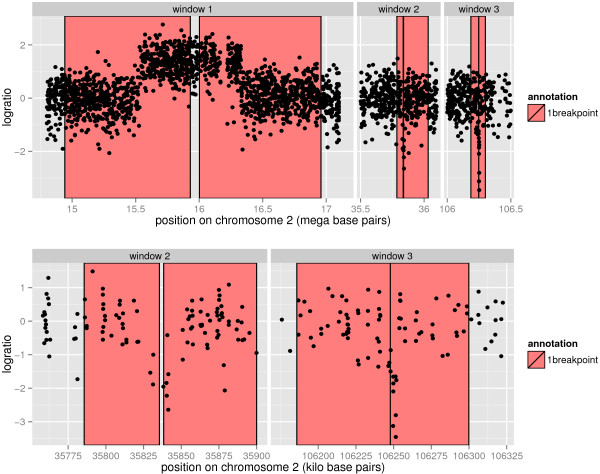
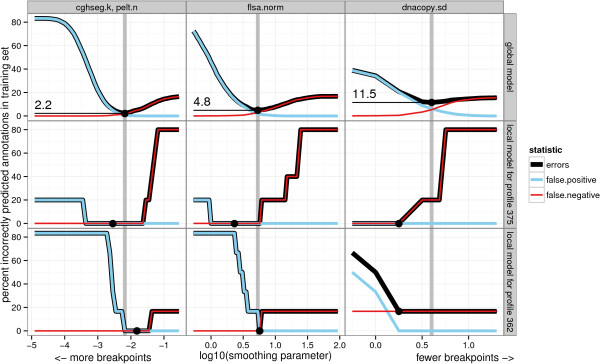
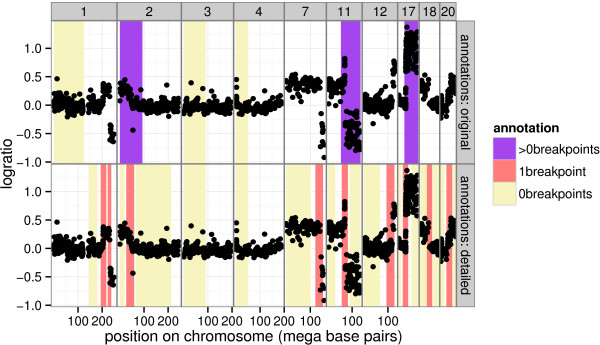
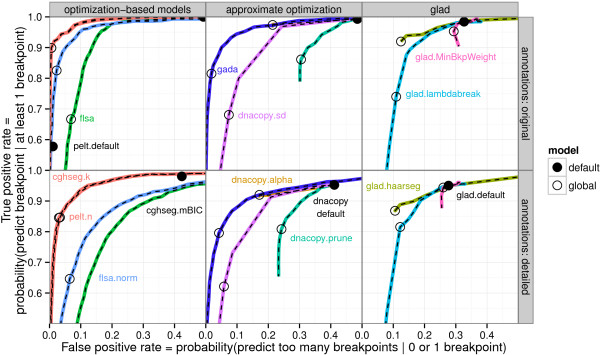
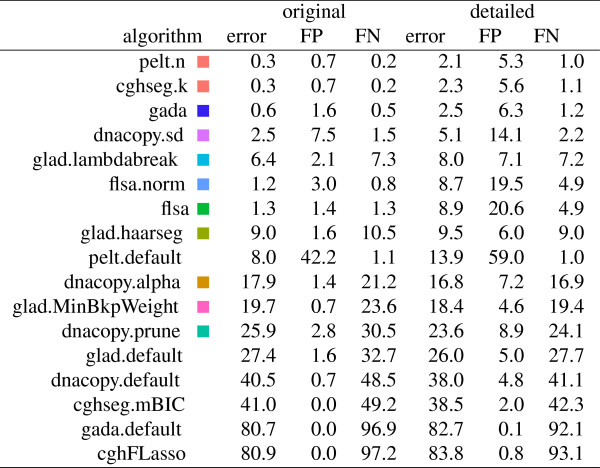
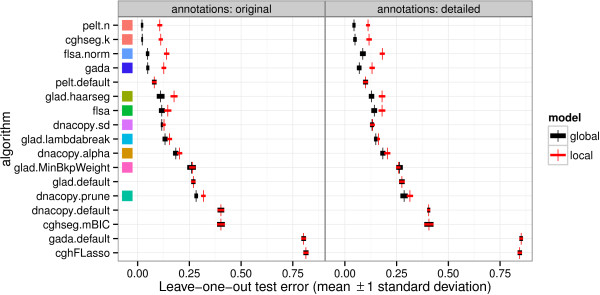
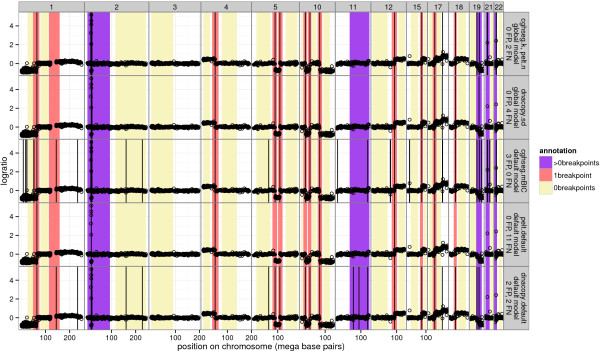
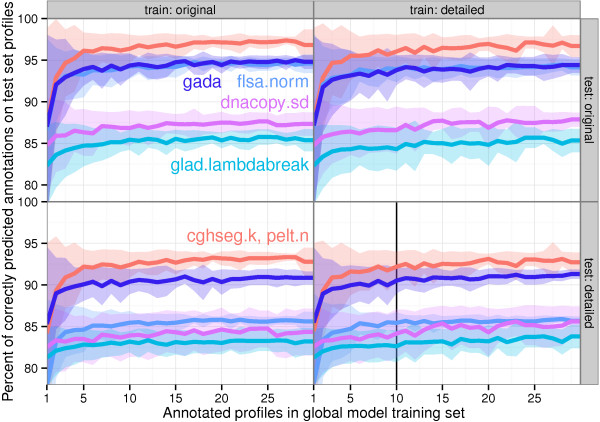
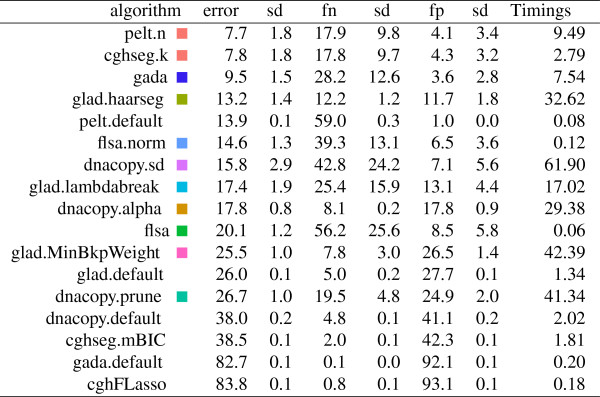
We present three contributions for copy number profile smoothing model selection. First, we propose to select the model and degree of smoothness that maximizes agreement with visual breakpoint region annotations. Second, we develop cross-validation procedures to estimate the error of the trained models. Third, we apply these methods to compare 17 smoothing models on a new database of 575 annotated neuroblastoma copy number profiles, which we make available as a public benchmark for testing new algorithms.
Whereas previous studies have been qualitative or limited to simulated data, our annotation-guided approach is quantitative and suggests which algorithms are fastest and most accurate in practice on real data. In the neuroblastoma data, the equivalent pelt.n and cghseg.k methods were the best breakpoint detectors, and exhibited reasonable computation times.
许多模型已被提出用于检测染色体拷贝数图谱中的拷贝数改变,但通常难以确定对于给定数据集哪种模型最有效。此外,大多数方法都具有一个平滑参数,用于确定断点的数量,并且必须使用各种启发式方法进行选择。
我们提出了三种用于拷贝数图谱平滑模型选择的方法。首先,我们提出选择与视觉断点区域注释最一致的模型和平滑程度。其次,我们开发了交叉验证程序来估计训练模型的误差。第三,我们应用这些方法在一个新的 575 个注释神经母细胞瘤拷贝数图谱的数据库上比较了 17 种平滑模型,我们将其作为一个公共基准,用于测试新算法。
虽然以前的研究是定性的或仅限于模拟数据,但我们的基于注释的方法是定量的,并建议哪些算法在实际的真实数据中最快和最准确。在神经母细胞瘤数据中,等效的 pelt.n 和 cghseg.k 方法是最好的断点检测方法,并且计算时间合理。