Hocking Toby Dylan, Goerner-Potvin Patricia, Morin Andreanne, Shao Xiaojian, Pastinen Tomi, Bourque Guillaume
Department of Human Genetics, McGill University, H3A-1A4, Montréal, Canada.
Bioinformatics. 2017 Feb 15;33(4):491-499. doi: 10.1093/bioinformatics/btw672.
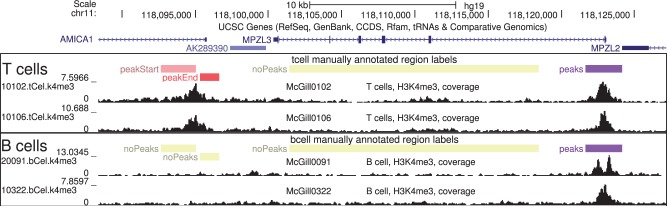
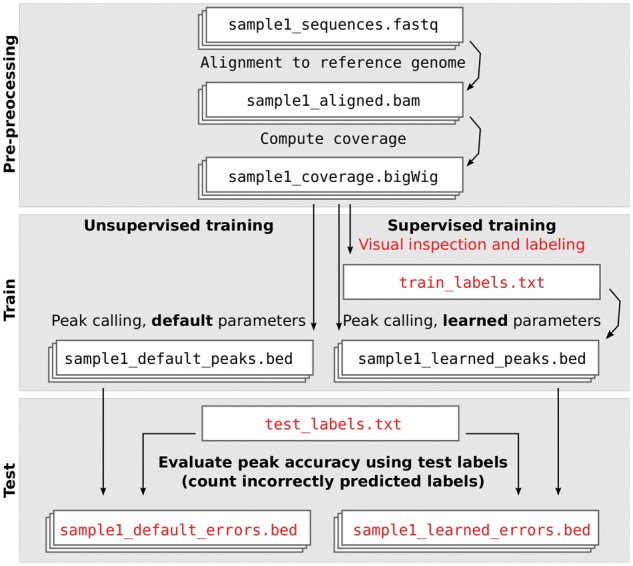
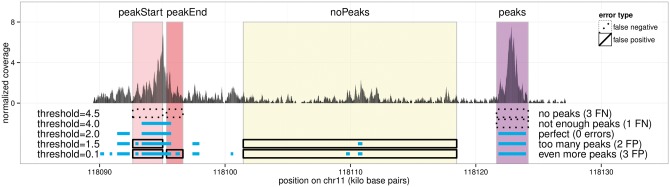
Many peak detection algorithms have been proposed for ChIP-seq data analysis, but it is not obvious which algorithm and what parameters are optimal for any given dataset. In contrast, regions with and without obvious peaks can be easily labeled by visual inspection of aligned read counts in a genome browser. We propose a supervised machine learning approach for ChIP-seq data analysis, using labels that encode qualitative judgments about which genomic regions contain or do not contain peaks. The main idea is to manually label a small subset of the genome, and then learn a model that makes consistent peak predictions on the rest of the genome.
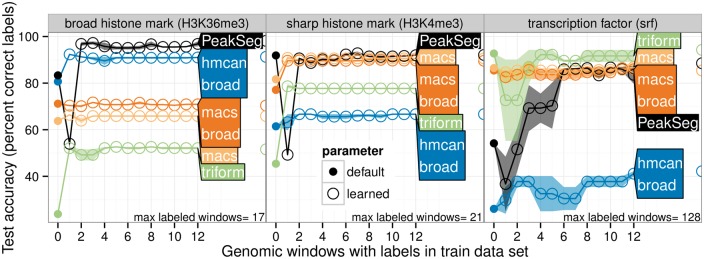
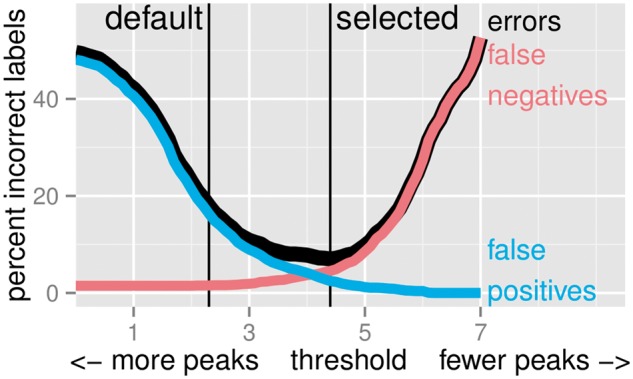
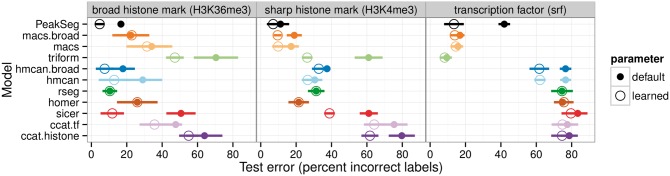
We created 7 new histone mark datasets with 12 826 visually determined labels, and analyzed 3 existing transcription factor datasets. We observed that default peak detection parameters yield high false positive rates, which can be reduced by learning parameters using a relatively small training set of labeled data from the same experiment type. We also observed that labels from different people are highly consistent. Overall, these data indicate that our supervised labeling method is useful for quantitatively training and testing peak detection algorithms.
Labeled histone mark data http://cbio.ensmp.fr/~thocking/chip-seq-chunk-db/ , R package to compute the label error of predicted peaks https://github.com/tdhock/PeakError.
toby.hocking@mail.mcgill.ca or guil.bourque@mcgill.ca.
Supplementary data are available at Bioinformatics online.
已经提出了许多用于ChIP-seq数据分析的峰值检测算法,但对于任何给定数据集,哪种算法和哪些参数是最优的并不明显。相比之下,通过在基因组浏览器中目视检查比对后的读数计数,可以轻松标记有明显峰值和无明显峰值的区域。我们提出了一种用于ChIP-seq数据分析的监督机器学习方法,使用对哪些基因组区域包含或不包含峰值进行定性判断编码的标签。主要思想是手动标记基因组的一小部分,然后学习一个能对基因组其余部分做出一致峰值预测的模型。
我们创建了7个新的组蛋白标记数据集,带有12826个通过目视确定的标签,并分析了3个现有的转录因子数据集。我们观察到默认的峰值检测参数会产生较高的假阳性率,通过使用来自相同实验类型的相对较小的标记数据训练集来学习参数,可以降低该比率。我们还观察到不同人员给出的标签高度一致。总体而言,这些数据表明我们的监督标记方法对于定量训练和测试峰值检测算法很有用。
toby.hocking@mail.mcgill.ca或guil.bourque@mcgill.ca。
补充数据可在《生物信息学》在线获取。