Department of Biology, University of Waterloo, Waterloo, ON, Canada.
Virulence. 2013 Aug 15;4(6):453-66. doi: 10.4161/viru.25180. Epub 2013 May 28.
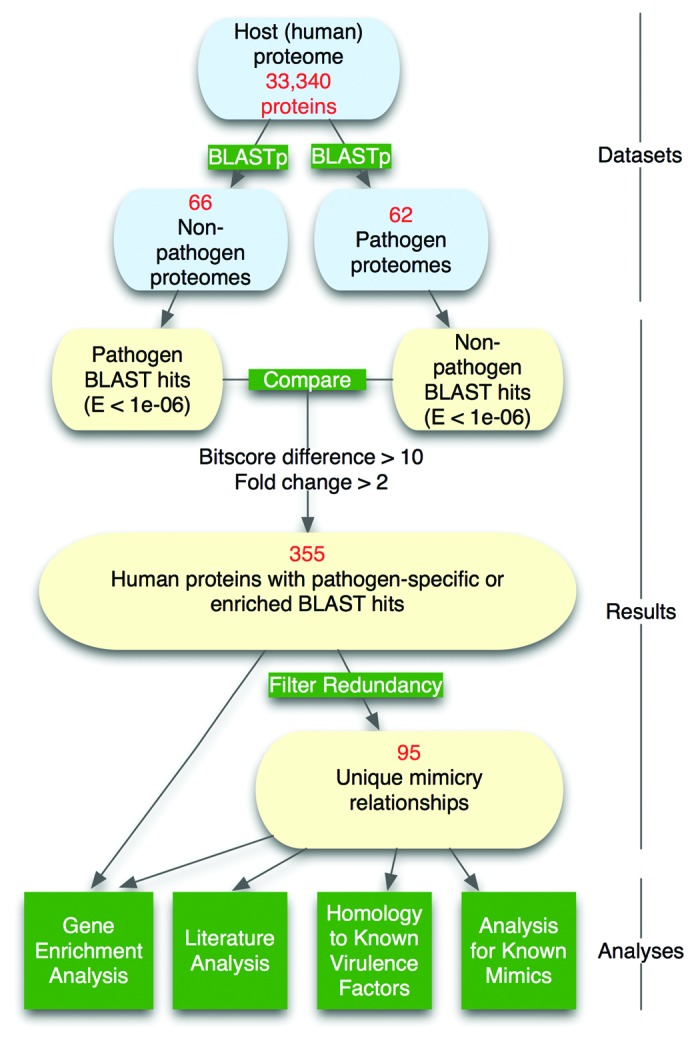
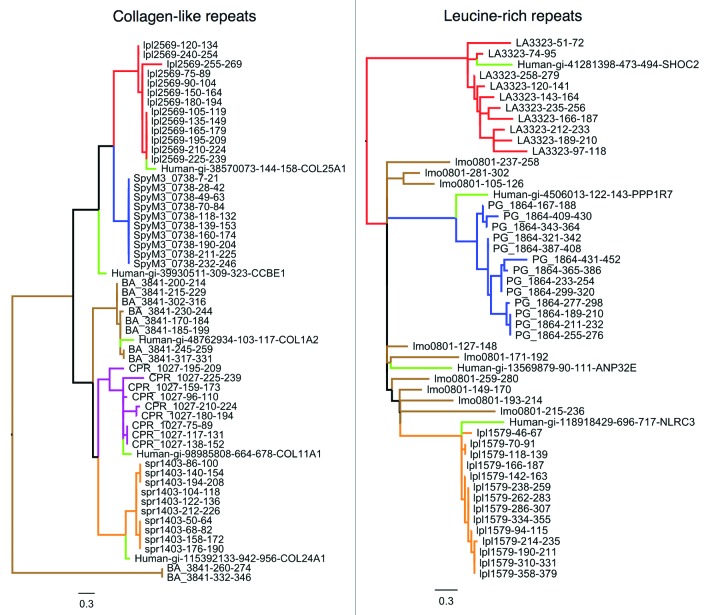
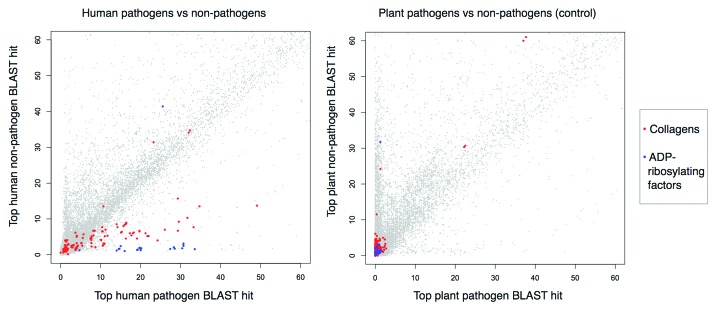
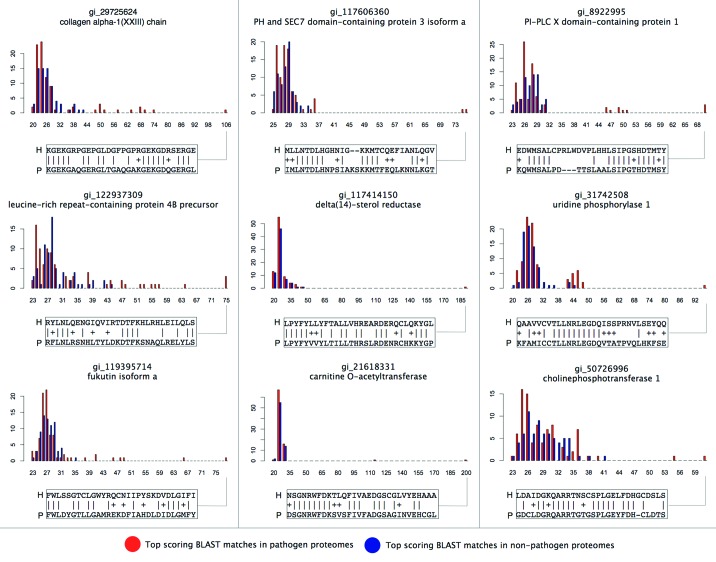
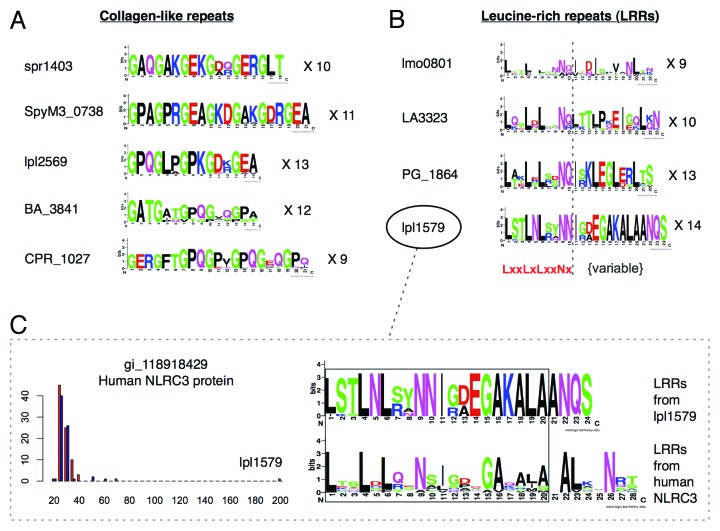
Molecular mimicry of host proteins is a common strategy adopted by bacterial pathogens to interfere with and exploit host processes. Despite the availability of pathogen genomes, few studies have attempted to predict virulence-associated mimicry relationships directly from genomic sequences. Here, we analyzed the proteomes of 62 pathogenic and 66 non-pathogenic bacterial species, and screened for the top pathogen-specific or pathogen-enriched sequence similarities to human proteins. The screen identified approximately 100 potential mimicry relationships including well-characterized examples among the top-scoring hits (e.g., RalF, internalin, yopH, and others), with about 1/3 of predicted relationships supported by existing literature. Examination of homology to virulence factors, statistically enriched functions, and comparison with literature indicated that the detected mimics target key host structures (e.g., extracellular matrix, ECM) and pathways (e.g., cell adhesion, lipid metabolism, and immune signaling). The top-scoring and most widespread mimicry pattern detected among pathogens consisted of elevated sequence similarities to ECM proteins including collagens and leucine-rich repeat proteins. Unexpectedly, analysis of the pathogen counterparts of these proteins revealed that they have evolved independently in different species of bacterial pathogens from separate repeat amplifications. Thus, our analysis provides evidence for two classes of mimics: complex proteins such as enzymes that have been acquired by eukaryote-to-pathogen horizontal transfer, and simpler repeat proteins that have independently evolved to mimic the host ECM. Ultimately, computational detection of pathogen-specific and pathogen-enriched similarities to host proteins provides insights into potentially novel mimicry-mediated virulence mechanisms of pathogenic bacteria.
宿主蛋白的分子模拟是细菌病原体用来干扰和利用宿主过程的常见策略。尽管病原体基因组已经存在,但很少有研究试图直接从基因组序列预测与毒力相关的模拟关系。在这里,我们分析了 62 种致病性和 66 种非致病性细菌物种的蛋白质组,并筛选了与人类蛋白质具有最高病原体特异性或病原体富集序列相似性的序列。该筛选确定了大约 100 种潜在的模拟关系,其中包括排名靠前的命中(例如,RalF、内毒素、yopH 等)中的一些特征明显的例子,大约 1/3 的预测关系得到了现有文献的支持。对同源性到毒力因子、统计上丰富的功能以及与文献的比较表明,检测到的模拟物针对关键的宿主结构(例如细胞外基质、ECM)和途径(例如细胞黏附、脂质代谢和免疫信号)。在病原体中检测到的得分最高和最广泛的模拟模式包括与 ECM 蛋白(包括胶原蛋白和富含亮氨酸的重复蛋白)的序列相似性升高。出乎意料的是,对这些蛋白质的病原体对应物的分析表明,它们在来自不同细菌病原体的不同物种中通过独立的重复扩增而进化。因此,我们的分析为两类模拟物提供了证据:复杂的蛋白质,如通过真核生物到病原体的水平转移获得的酶,以及更简单的重复蛋白质,它们独立进化以模拟宿主 ECM。最终,计算检测病原体特异性和病原体富集与宿主蛋白的相似性为潜在的新型模拟介导的致病菌毒力机制提供了深入的了解。