Zhang Yang
Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan, 48109; Department of Biological Chemistry, University of Michigan, Ann Arbor, Michigan, 48109.
Proteins. 2014 Feb;82 Suppl 2(0 2):175-87. doi: 10.1002/prot.24341. Epub 2013 Aug 31.
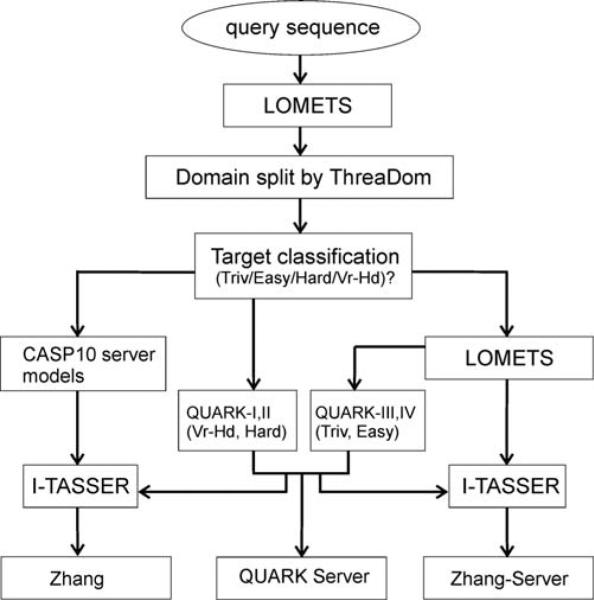
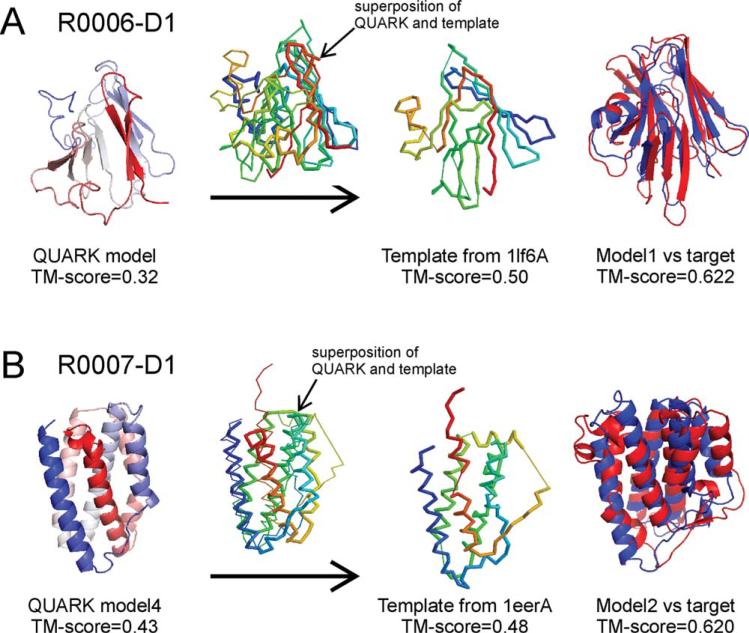
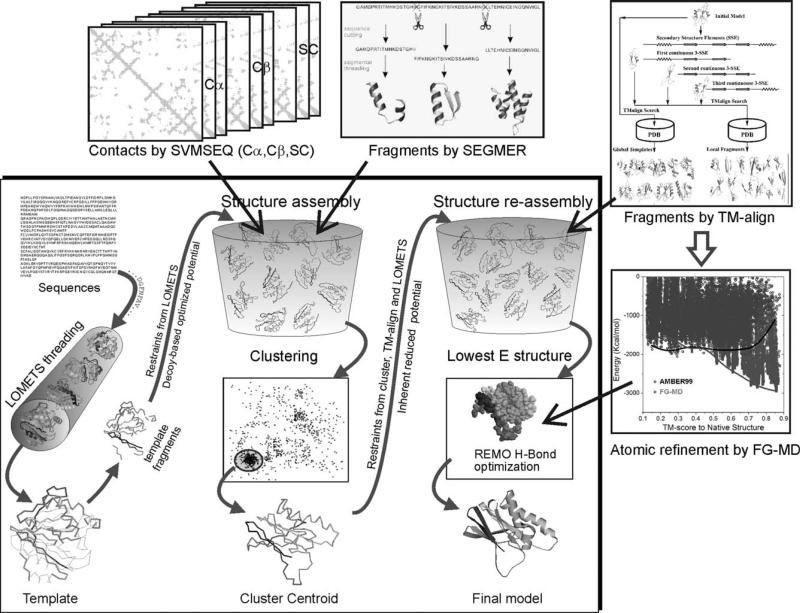
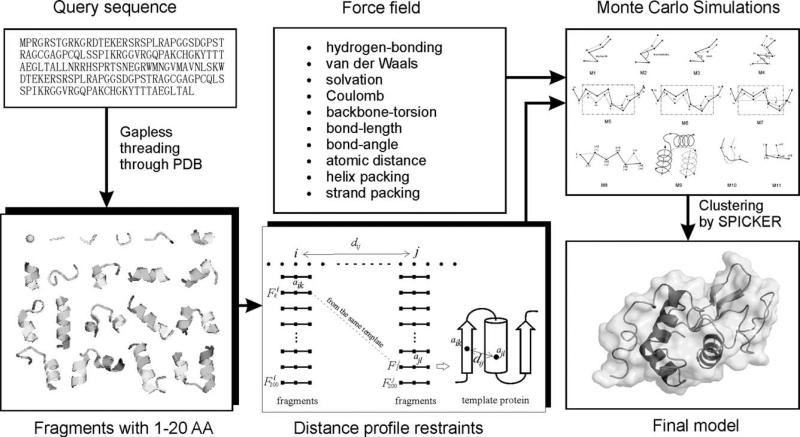
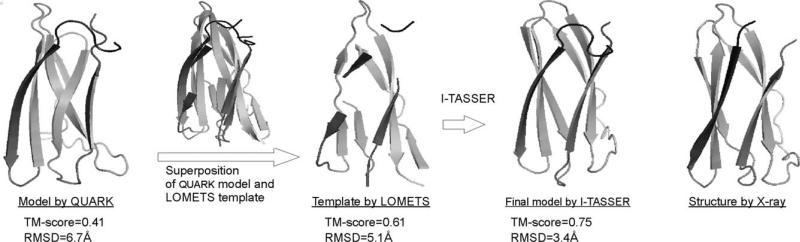
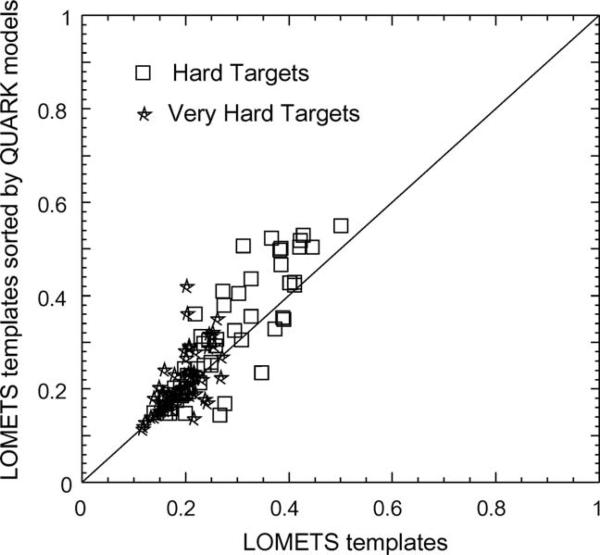
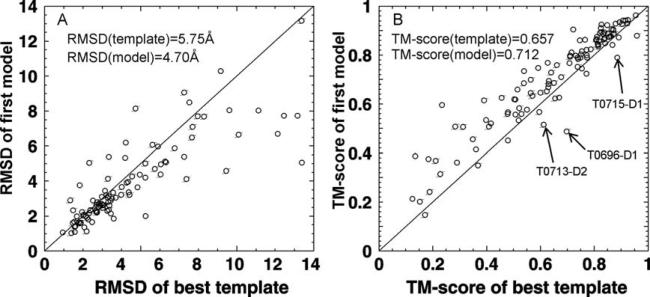
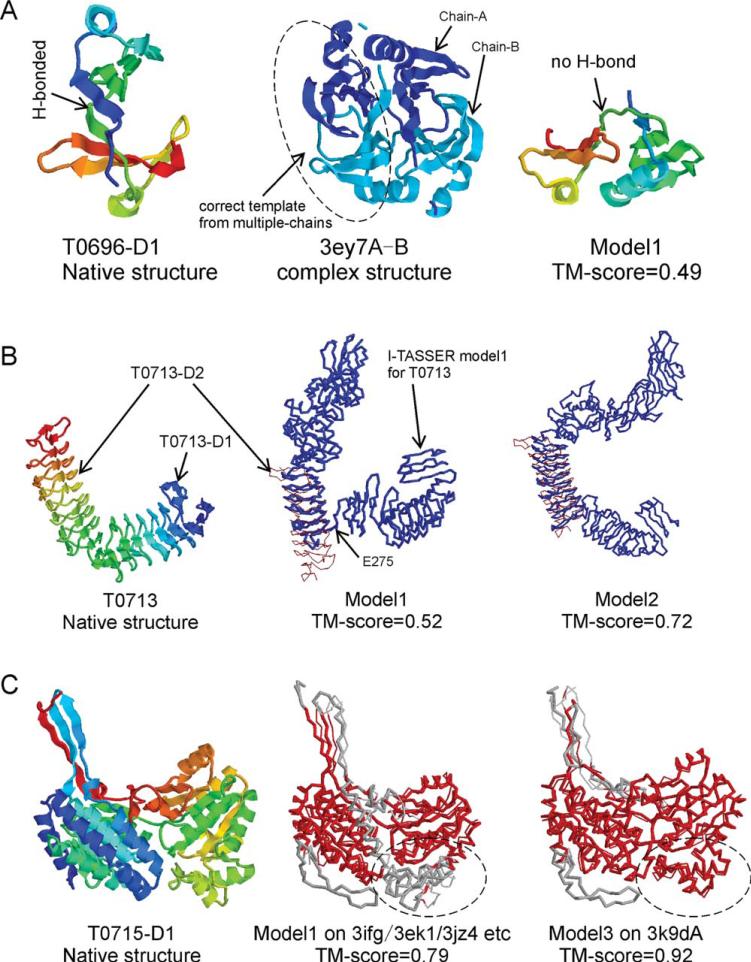
We develop and test a new pipeline in CASP10 to predict protein structures based on an interplay of I-TASSER and QUARK for both free-modeling (FM) and template-based modeling (TBM) targets. The most noteworthy observation is that sorting through the threading template pool using the QUARK-based ab initio models as probes allows the detection of distant-homology templates which might be ignored by the traditional sequence profile-based threading alignment algorithms. Further template assembly refinement by I-TASSER resulted in successful folding of two medium-sized FM targets with >150 residues. For TBM, the multiple threading alignments from LOMETS are, for the first time, incorporated into the ab initio QUARK simulations, which were further refined by I-TASSER assembly refinement. Compared with the traditional threading assembly refinement procedures, the inclusion of the threading-constrained ab initio folding models can consistently improve the quality of the full-length models as assessed by the GDT-HA and hydrogen-bonding scores. Despite the success, significant challenges still exist in domain boundary prediction and consistent folding of medium-size proteins (especially beta-proteins) for nonhomologous targets. Further developments of sensitive fold-recognition and ab initio folding methods are critical for solving these problems.
我们在蛋白质结构预测技术关键评估第10轮(CASP10)中开发并测试了一种新的流程,该流程基于I-TASSER和QUARK的相互作用,对自由建模(FM)和基于模板的建模(TBM)目标进行蛋白质结构预测。最值得注意的发现是,使用基于QUARK的从头算模型作为探针在穿线模板库中进行筛选,可以检测到传统的基于序列谱的穿线比对算法可能忽略的远同源模板。通过I-TASSER进一步进行模板组装优化,成功折叠了两个含有超过150个残基的中等大小的FM目标。对于TBM,首次将来自LOMETS的多穿线比对纳入从头算QUARK模拟中,并通过I-TASSER组装优化进一步完善。与传统的穿线组装优化程序相比,纳入受穿线约束的从头算折叠模型,根据全局距离测试-全原子(GDT-HA)和氢键得分评估,能够持续提高全长模型的质量。尽管取得了成功,但在非同源目标的结构域边界预测和中等大小蛋白质(尤其是β-蛋白质)的一致折叠方面,仍然存在重大挑战。进一步开发灵敏的折叠识别和从头算折叠方法对于解决这些问题至关重要。