James Geo Velikkakam, Patel Vipul, Nordström Karl J V, Klasen Jonas R, Salomé Patrice A, Weigel Detlef, Schneeberger Korbinian
Genome Biol. 2013 Jun 17;14(6):R61. doi: 10.1186/gb-2013-14-6-r61.
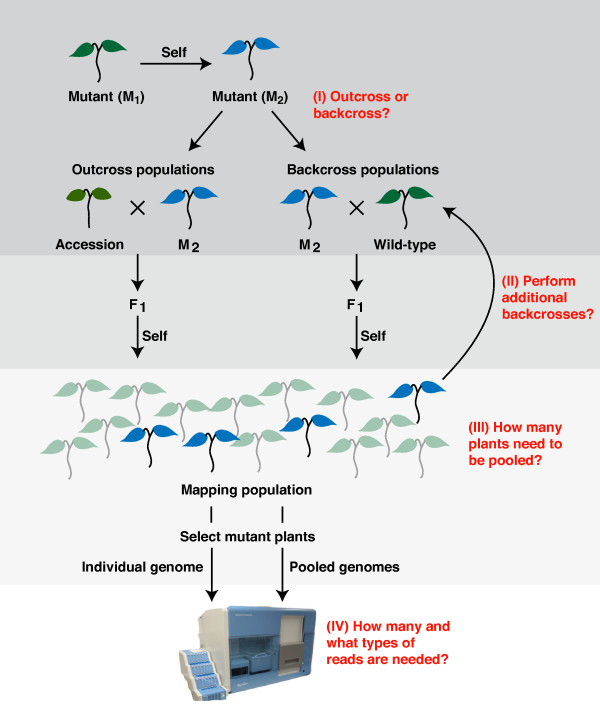
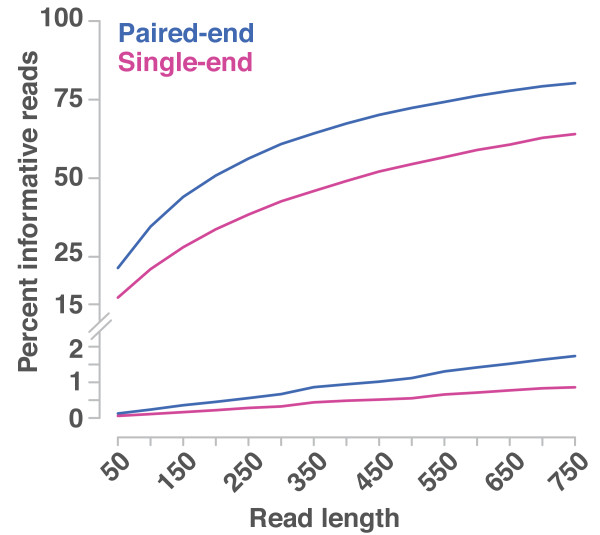
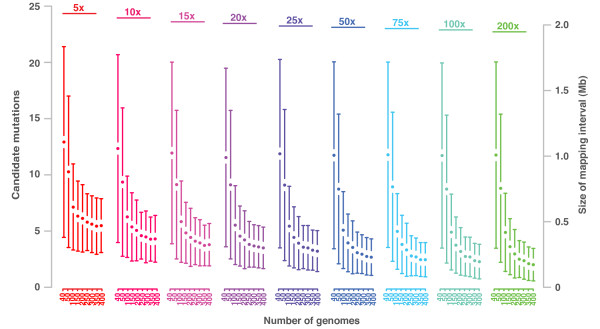
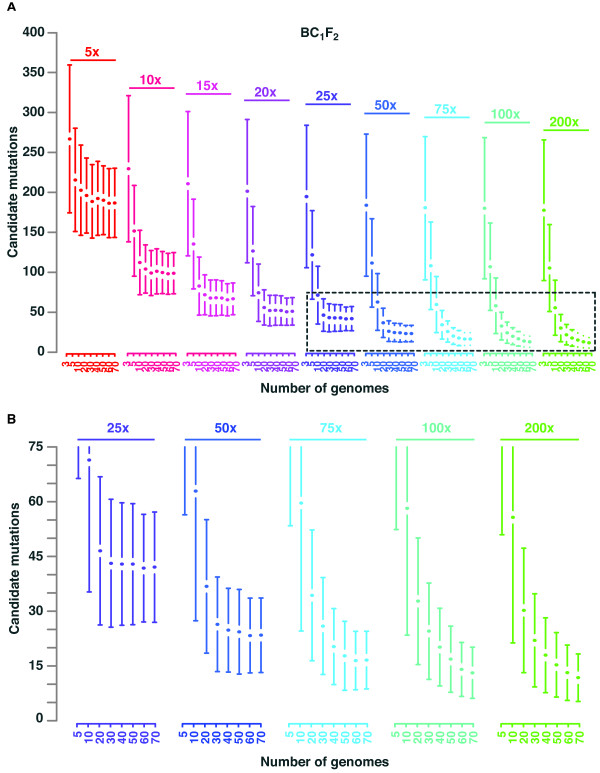
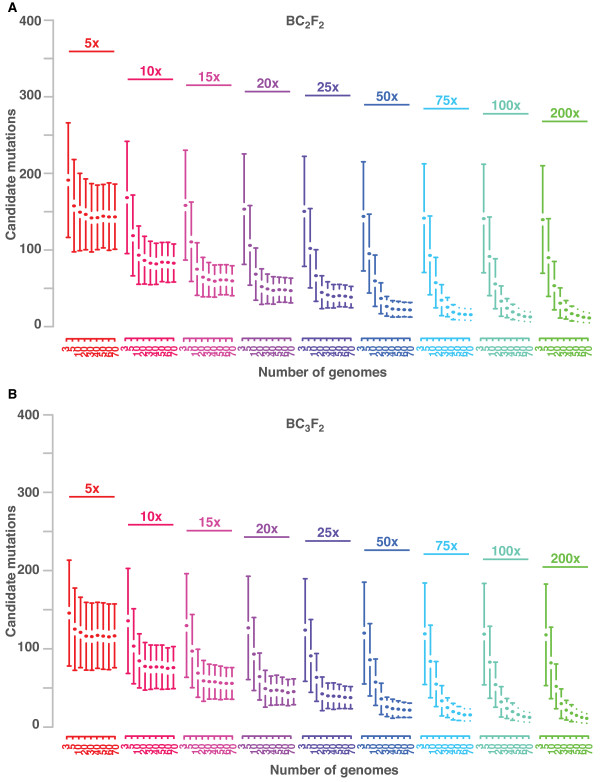
Mapping-by-sequencing combines genetic mapping with whole-genome sequencing in order to accelerate mutant identification. However, application of mapping-by-sequencing requires decisions on various practical settings on the experimental design that are not intuitively answered. Following an experimentally determined recombination landscape of Arabidopsis and next generation sequencing-specific biases, we simulated more than 400,000 mapping-by-sequencing experiments. This allowed us to evaluate a broad range of different types of experiments and to develop general rules for mapping-by-sequencing in Arabidopsis. Most importantly, this informs about the properties of different crossing scenarios, the number of recombinants and sequencing depth needed for successful mapping experiments.
测序定位将遗传定位与全基因组测序相结合,以加速突变体鉴定。然而,测序定位的应用需要对实验设计中的各种实际设置做出决策,而这些问题并没有直观的答案。根据实验确定的拟南芥重组图谱和新一代测序特有的偏差,我们模拟了超过400,000次测序定位实验。这使我们能够评估广泛的不同类型实验,并制定拟南芥测序定位的通用规则。最重要的是,这为不同杂交方案的特性、成功定位实验所需的重组体数量和测序深度提供了信息。