Department of Computer Science, Duke University, Durham, NC 27708, USA.
Bioinformatics. 2013 Jul 1;29(13):i108-16. doi: 10.1093/bioinformatics/btt233.
Pre-mRNA cleavage and polyadenylation are essential steps for 3'-end maturation and subsequent stability and degradation of mRNAs. This process is highly controlled by cis-regulatory elements surrounding the cleavage/polyadenylation sites (polyA sites), which are frequently constrained by sequence content and position. More than 50% of human transcripts have multiple functional polyA sites, and the specific use of alternative polyA sites (APA) results in isoforms with variable 3'-untranslated regions, thus potentially affecting gene regulation. Elucidating the regulatory mechanisms underlying differential polyA preferences in multiple cell types has been hindered both by the lack of suitable data on the precise location of cleavage sites, as well as of appropriate tests for determining APAs with significant differences across multiple libraries.
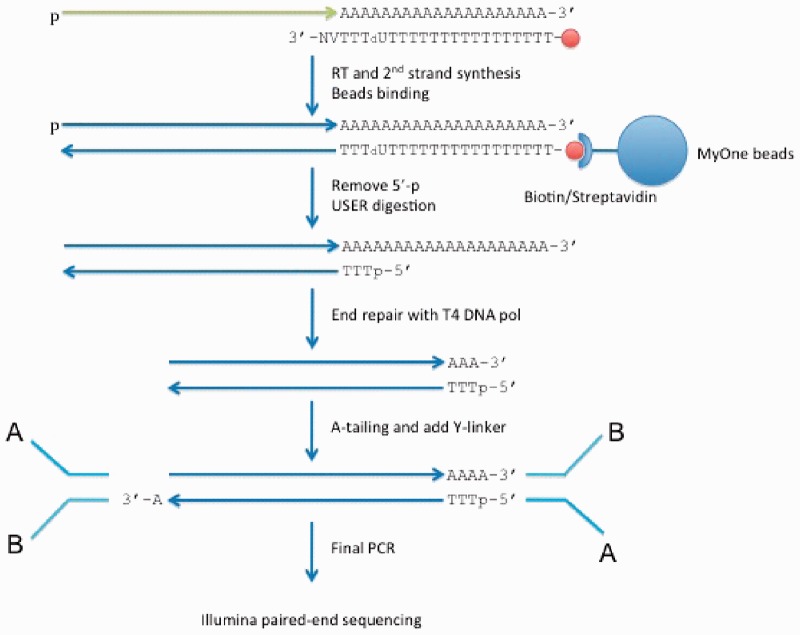
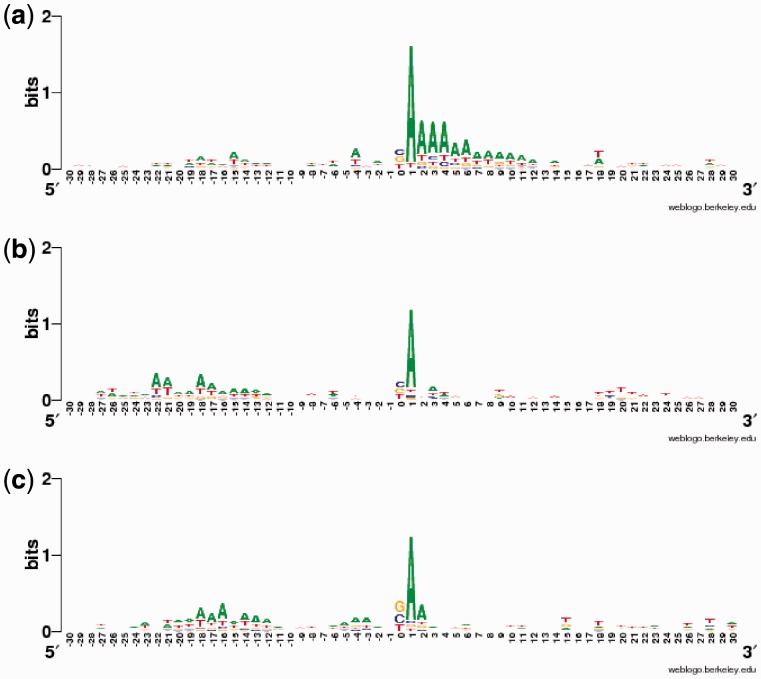
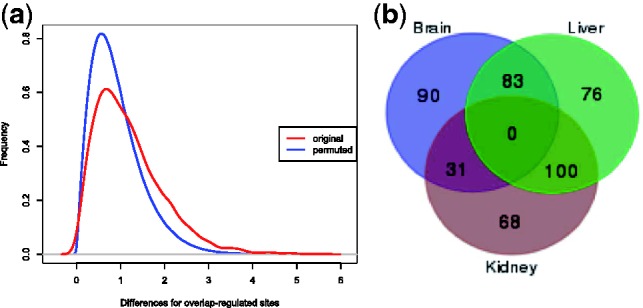
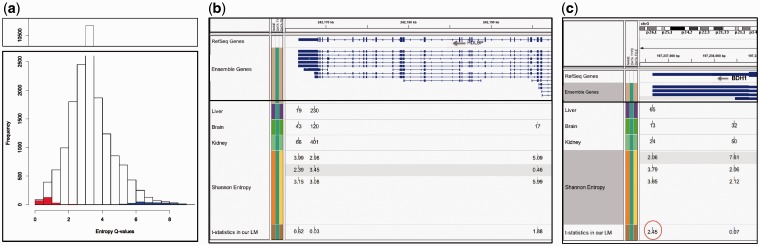
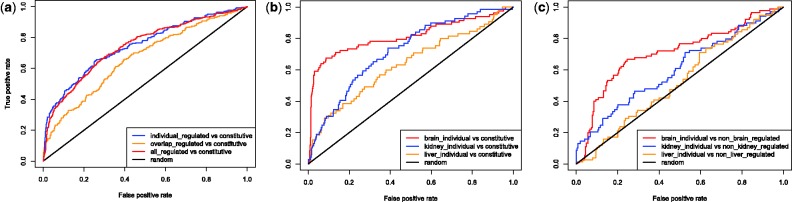
We applied a tailored paired-end RNA-seq protocol to specifically probe the position of polyA sites in three human adult tissue types. We specified a linear-effects regression model to identify tissue-specific biases indicating regulated APA; the significance of differences between tissue types was assessed by an appropriately designed permutation test. This combination allowed to identify highly specific subsets of APA events in the individual tissue types. Predictive models successfully classified constitutive polyA sites from a biologically relevant background (auROC = 99.6%), as well as tissue-specific regulated sets from each other. We found that the main cis-regulatory elements described for polyadenylation are a strong, and highly informative, hallmark for constitutive sites only. Tissue-specific regulated sites were found to contain other regulatory motifs, with the canonical polyadenylation signal being nearly absent at brain-specific polyA sites. Together, our results contribute to the understanding of the diversity of post-transcriptional gene regulation.
Raw data are deposited on SRA, accession numbers: brain SRX208132, kidney SRX208087 and liver SRX208134. Processed datasets as well as model code are published on our website: http://www.genome.duke.edu/labs/ohler/research/UTR/.
前体 mRNA 的切割和多聚腺苷酸化是 3'端成熟以及随后 mRNA 的稳定性和降解所必需的步骤。这个过程受到切割/多聚腺苷酸化位点(多聚 A 位点)周围顺式调控元件的高度调控,这些元件经常受到序列内容和位置的限制。超过 50%的人类转录本具有多个功能多聚 A 位点,而替代多聚 A 位点(APA)的特异性使用导致具有可变 3'非翻译区的异构体,从而可能影响基因调控。阐明多种细胞类型中不同多聚 A 偏好的调控机制,既受到缺乏关于切割位点精确位置的合适数据的限制,也受到缺乏在多个文库中具有显著差异的 APA 的适当测试的限制。
我们应用了一种定制的配对末端 RNA-seq 方案,专门探测三个人类成年组织类型中多聚 A 位点的位置。我们指定了一个线性效应回归模型来识别指示调节 APA 的组织特异性偏差;通过适当设计的置换检验来评估组织类型之间差异的显著性。这种组合允许在单个组织类型中识别出高度特异性的 APA 事件子集。预测模型成功地将组成型多聚 A 位点从生物学相关背景中分类(auROC = 99.6%),并将彼此的组织特异性调节集分类。我们发现,多聚腺苷酸化描述的主要顺式调控元件是组成型位点的一个强有力且高度信息丰富的特征。组织特异性调节位点包含其他调控基序,而在脑特异性多聚 A 位点中几乎不存在典型的多聚腺苷酸化信号。总之,我们的结果有助于理解转录后基因调控的多样性。
原始数据已存储在 SRA 中, accession numbers:brain SRX208132,kidney SRX208087 和 liver SRX208134。处理后的数据集以及模型代码已发布在我们的网站上:http://www.genome.duke.edu/labs/ohler/research/UTR/。