Integrative Systems Biology Laboratory, Biological and Environmental Sciences and Engineering Division, Computational Bioscience Research Center, King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia.
Bioinformatics. 2013 Jul 1;29(13):i199-209. doi: 10.1093/bioinformatics/btt208.
Most functions within the cell emerge thanks to protein-protein interactions (PPIs), yet experimental determination of PPIs is both expensive and time-consuming. PPI networks present significant levels of noise and incompleteness. Predicting interactions using only PPI-network topology (topological prediction) is difficult but essential when prior biological knowledge is absent or unreliable.
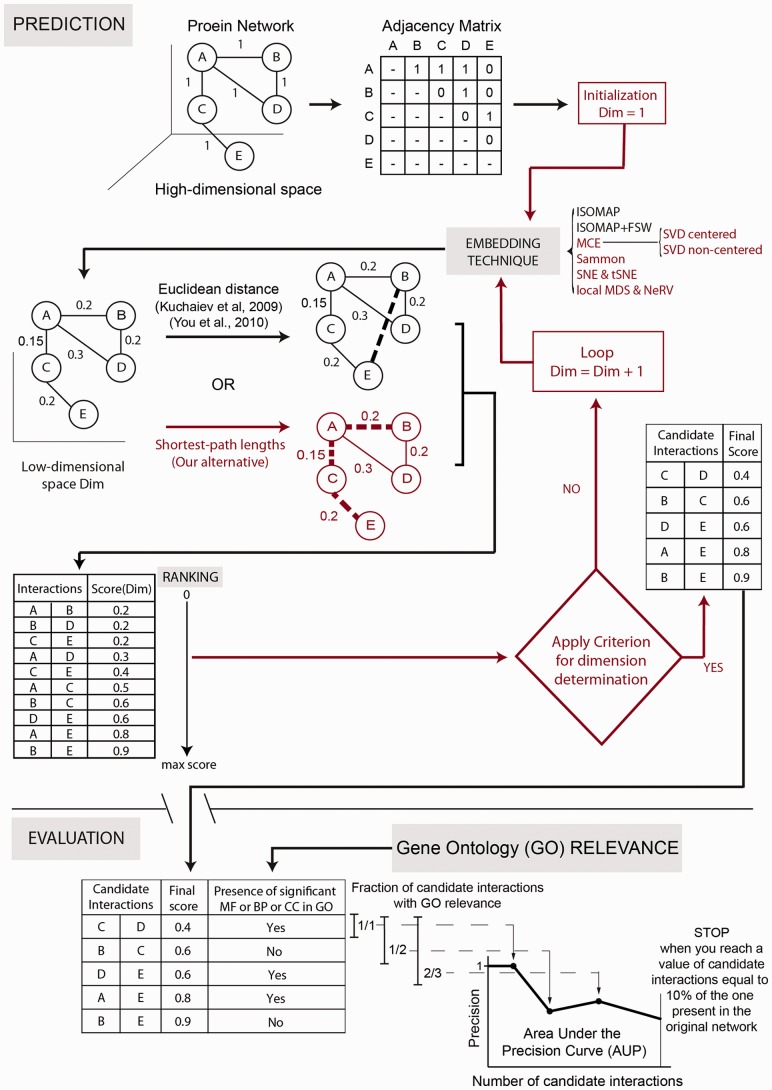
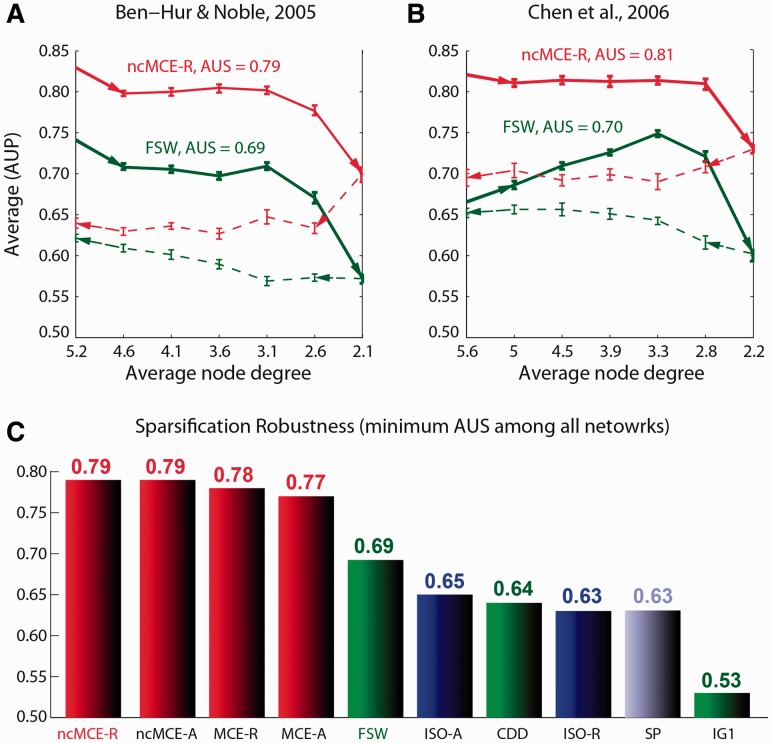
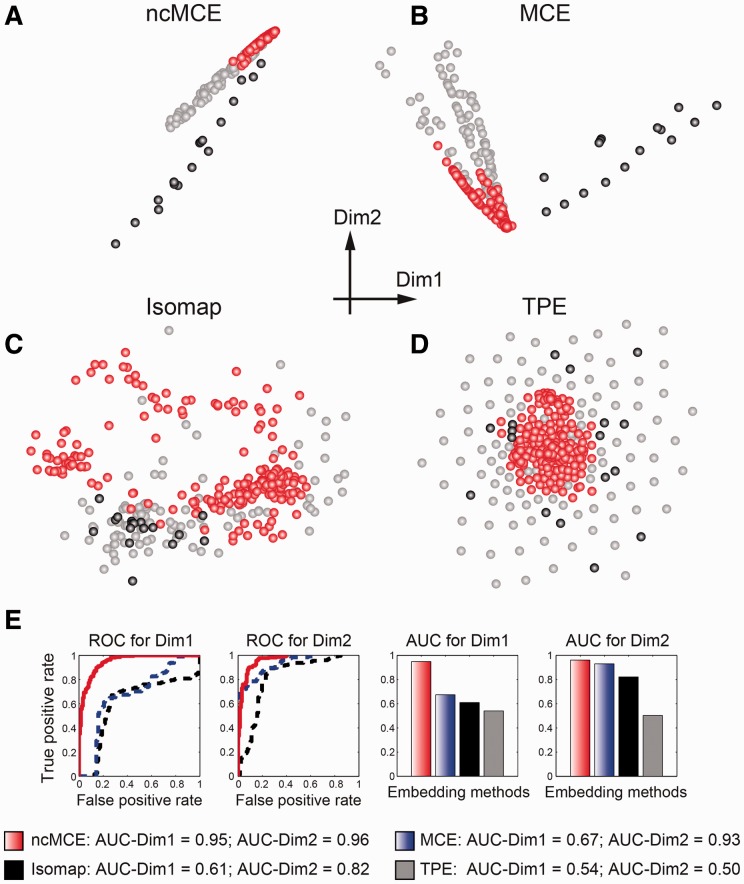
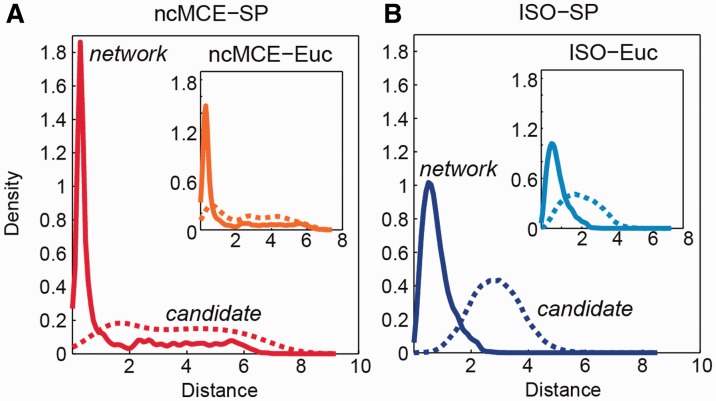
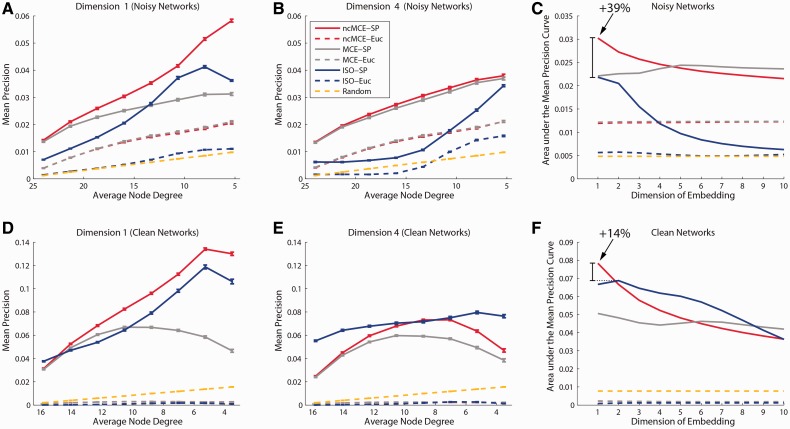
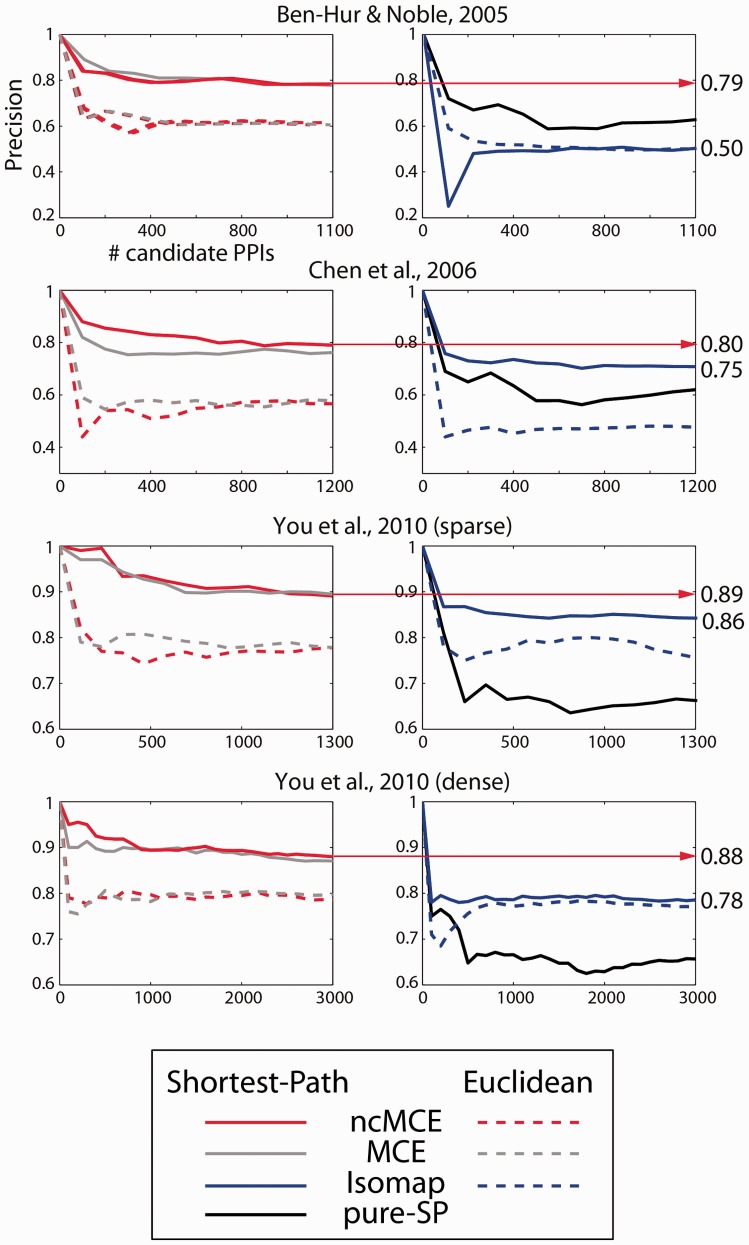
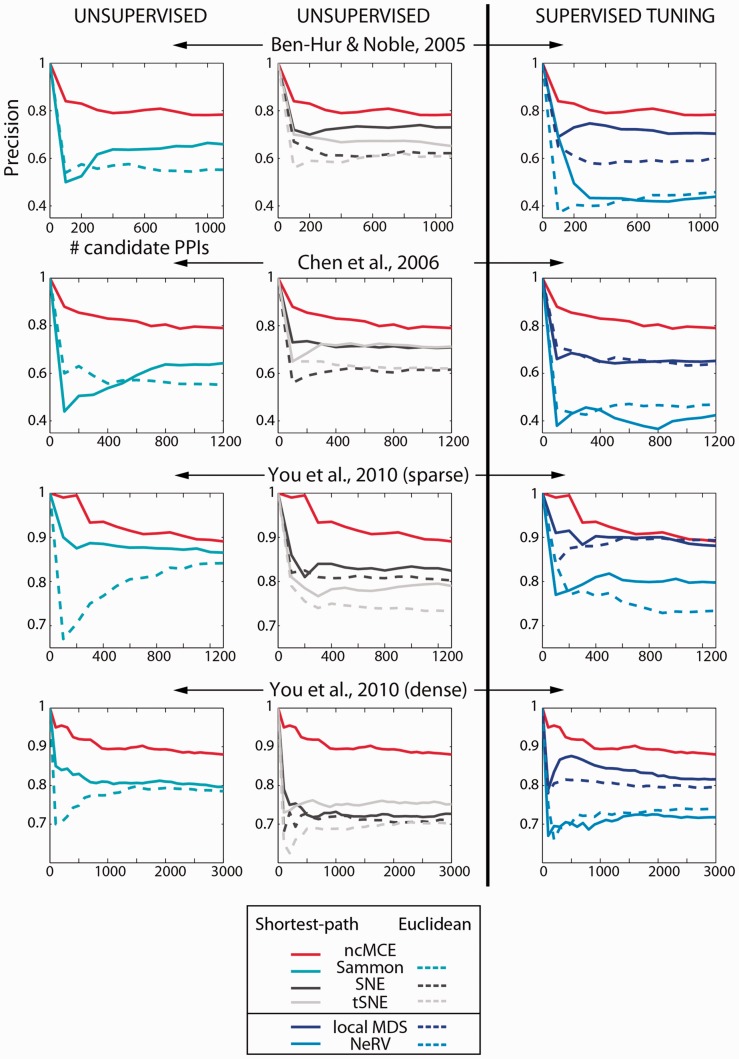
Network embedding emphasizes the relations between network proteins embedded in a low-dimensional space, in which protein pairs that are closer to each other represent good candidate interactions. To achieve network denoising, which boosts prediction performance, we first applied minimum curvilinear embedding (MCE), and then adopted shortest path (SP) in the reduced space to assign likelihood scores to candidate interactions. Furthermore, we introduce (i) a new valid variation of MCE, named non-centred MCE (ncMCE); (ii) two automatic strategies for selecting the appropriate embedding dimension; and (iii) two new randomized procedures for evaluating predictions.
We compared our method against several unsupervised and supervisedly tuned embedding approaches and node neighbourhood techniques. Despite its computational simplicity, ncMCE-SP was the overall leader, outperforming the current methods in topological link prediction.
Minimum curvilinearity is a valuable non-linear framework that we successfully applied to the embedding of protein networks for the unsupervised prediction of novel PPIs. The rationale for our approach is that biological and evolutionary information is imprinted in the non-linear patterns hidden behind the protein network topology, and can be exploited for predicting new protein links. The predicted PPIs represent good candidates for testing in high-throughput experiments or for exploitation in systems biology tools such as those used for network-based inference and prediction of disease-related functional modules.
https://sites.google.com/site/carlovittoriocannistraci/home.
Supplementary data are available at Bioinformatics online.
大多数细胞内的功能都是由于蛋白质-蛋白质相互作用(PPIs)而产生的,但实验确定 PPIs 既昂贵又耗时。PPI 网络存在显著的噪声和不完整性。仅使用 PPI 网络拓扑结构(拓扑预测)预测相互作用是困难的,但在缺乏或不可靠的先验生物学知识时是必不可少的。
网络嵌入强调嵌入在低维空间中的网络蛋白质之间的关系,其中彼此更接近的蛋白质对表示良好的候选相互作用。为了实现网络去噪,从而提高预测性能,我们首先应用最小曲率嵌入(MCE),然后在降维空间中采用最短路径(SP)为候选相互作用分配可能性得分。此外,我们引入了(i)MCE 的一种新的有效变体,称为非中心 MCE(ncMCE);(ii)两种用于选择适当嵌入维度的自动策略;以及(iii)两种用于评估预测的新随机程序。
我们将我们的方法与几种无监督和监督调整的嵌入方法和节点邻域技术进行了比较。尽管计算简单,但 ncMCE-SP 总体上处于领先地位,在拓扑链路预测方面优于当前方法。
最小曲率是一个有价值的非线性框架,我们成功地将其应用于蛋白质网络的嵌入,用于无监督预测新的 PPIs。我们方法的原理是,生物和进化信息印在蛋白质网络拓扑背后隐藏的非线性模式中,可以用于预测新的蛋白质链接。预测的 PPIs 是在高通量实验中进行测试或在系统生物学工具中利用的良好候选者,例如用于基于网络的推断和预测与疾病相关的功能模块的工具。
https://sites.google.com/site/carlovittoriocannistraci/home。
补充数据可在 Bioinformatics 在线获取。