State Key Laboratory of Natural and Biomimetic Drugs, Department of Pharmacology, School of Basic Medical Sciences, Peking University and Beijing Key Laboratory of Tumor Systems Biology, Peking University, Beijing, China.
PLoS One. 2013 Jun 25;8(6):e67142. doi: 10.1371/journal.pone.0067142. Print 2013.
Ulcerative colitis (UC) was the most frequently diagnosed inflammatory bowel disease (IBD) and closely linked to colorectal carcinogenesis. By far, the underlying mechanisms associated with the disease are still unclear. With the increasing accumulation of microarray gene expression profiles, it is profitable to gain a systematic perspective based on gene regulatory networks to better elucidate the roles of genes associated with disorders. However, a major challenge for microarray data analysis is the integration of multiple-studies generated by different groups.
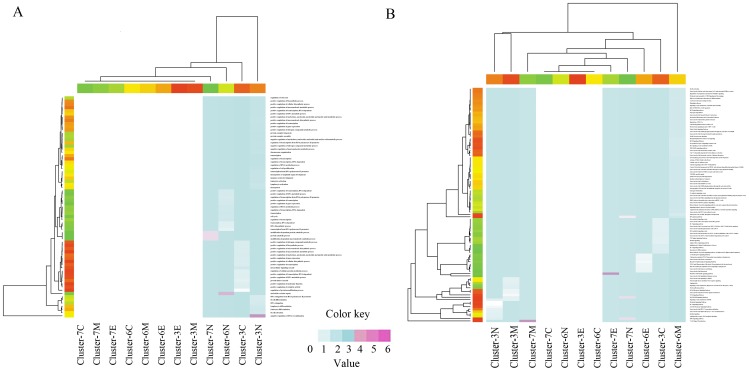
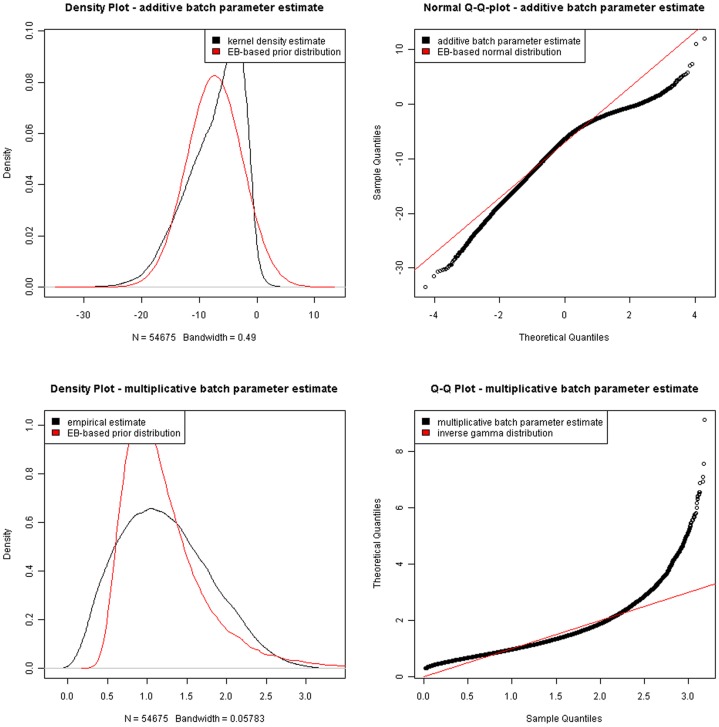
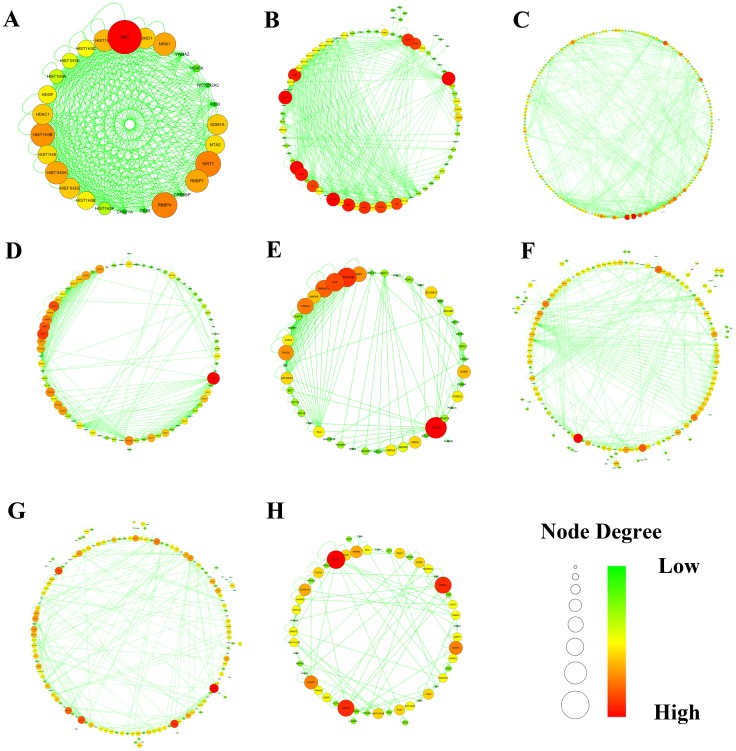
METHODOLOGY/PRINCIPAL FINDINGS: In this study, firstly, we modeled a signaling regulatory network associated with colorectal cancer (CRC) initiation via integration of cross-study microarray expression data sets using Empirical Bayes (EB) algorithm. Secondly, a manually curated human cancer signaling map was established via comprehensive retrieval of the publicly available repositories. Finally, the co-differently-expressed genes were manually curated to portray the layered signaling regulatory networks.
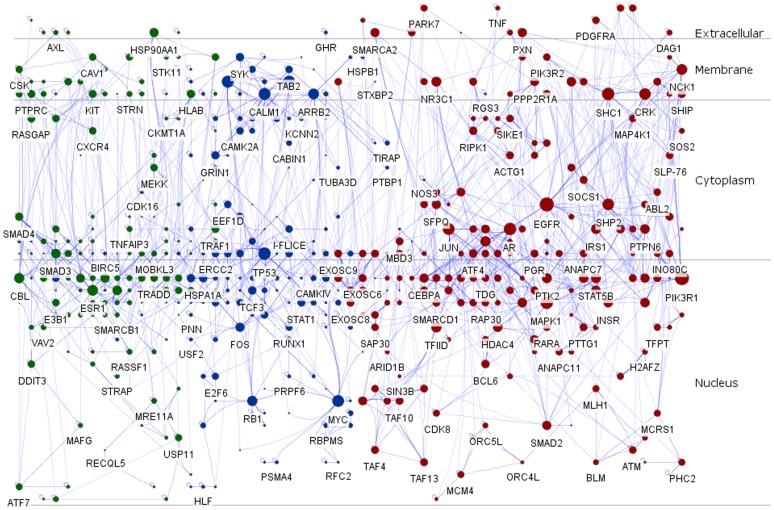
Overall, the remodeled signaling regulatory networks were separated into four major layers including extracellular, membrane, cytoplasm and nucleus, which led to the identification of five core biological processes and four signaling pathways associated with colorectal carcinogenesis. As a result, our biological interpretation highlighted the importance of EGF/EGFR signaling pathway, EPO signaling pathway, T cell signal transduction and members of the BCR signaling pathway, which were responsible for the malignant transition of CRC from the benign UC to the aggressive one.
The present study illustrated a standardized normalization approach for cross-study microarray expression data sets. Our model for signaling networks construction was based on the experimentally-supported interaction and microarray co-expression modeling. Pathway-based signaling regulatory networks analysis sketched a directive insight into colorectal carcinogenesis, which was of significant importance to monitor disease progression and improve therapeutic interventions.
溃疡性结肠炎(UC)是最常见的炎症性肠病(IBD)诊断,与结直肠癌的发生密切相关。到目前为止,与该疾病相关的潜在机制仍不清楚。随着基因表达谱微阵列的积累,基于基因调控网络获得系统的观点来更好地阐明与疾病相关的基因的作用是有利的。然而,微阵列数据分析的一个主要挑战是整合由不同组生成的多个研究。
方法/主要发现:在这项研究中,首先,我们通过使用经验贝叶斯(EB)算法整合跨研究微阵列表达数据集,对与结直肠癌(CRC)起始相关的信号转导网络进行建模。其次,通过全面检索公开可用的存储库,建立了人工 curated 的人类癌症信号图。最后,对差异表达基因进行了人工整理,以描绘分层信号转导网络。
总的来说,重构的信号转导网络分为四个主要层,包括细胞外、膜、细胞质和细胞核,这导致确定了与结直肠发生相关的五个核心生物学过程和四个信号通路。结果,我们的生物学解释强调了 EGF/EGFR 信号通路、EPO 信号通路、T 细胞信号转导和 BCR 信号通路成员的重要性,这些通路负责 CRC 从良性 UC 向侵袭性转变的恶性转化。
本研究说明了一种标准化的跨研究微阵列表达数据集归一化方法。我们的信号网络构建模型是基于实验支持的相互作用和微阵列共表达建模。基于途径的信号转导网络分析描绘了结直肠发生的直接见解,这对于监测疾病进展和改善治疗干预非常重要。