School of Mathematics and Statistics, University of New South Wales, Sydney, NSW, Australia.
PLoS One. 2013 Jun 20;8(6):e67254. doi: 10.1371/journal.pone.0067254. Print 2013.
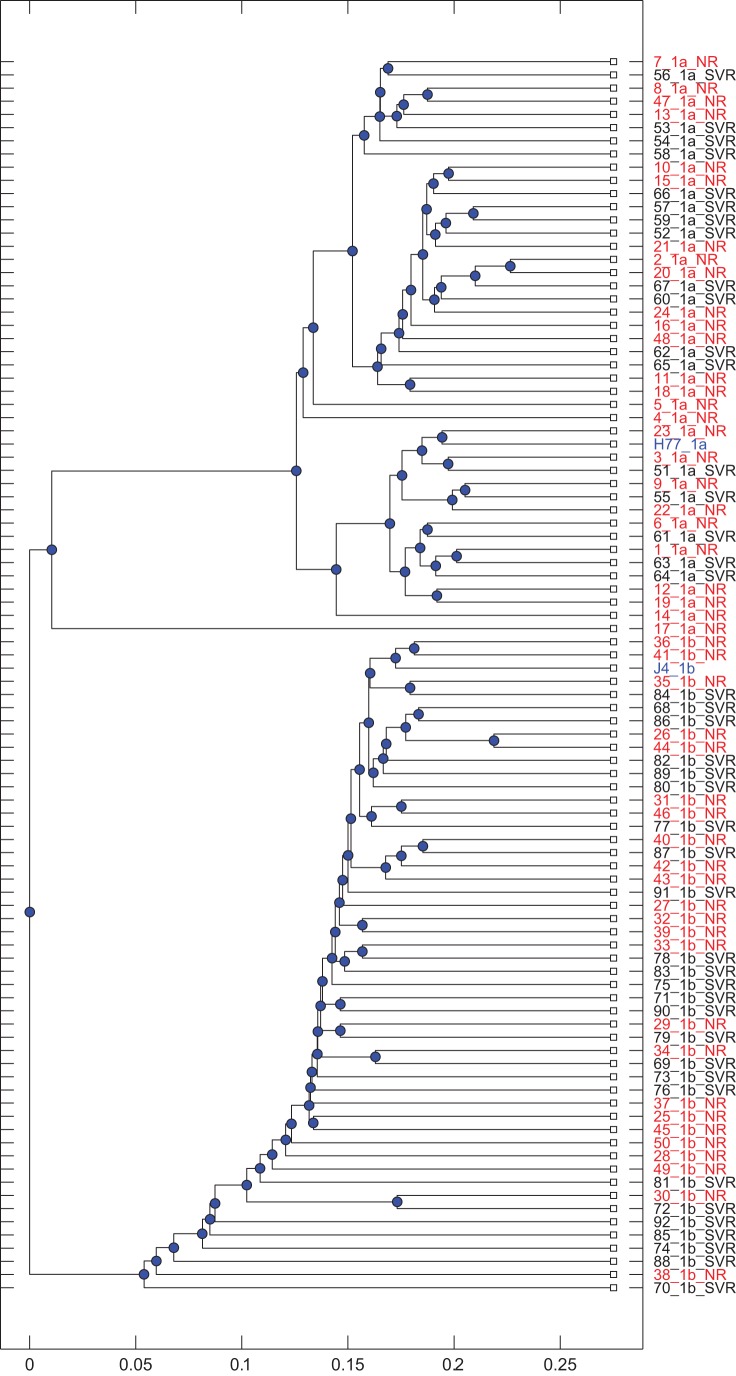
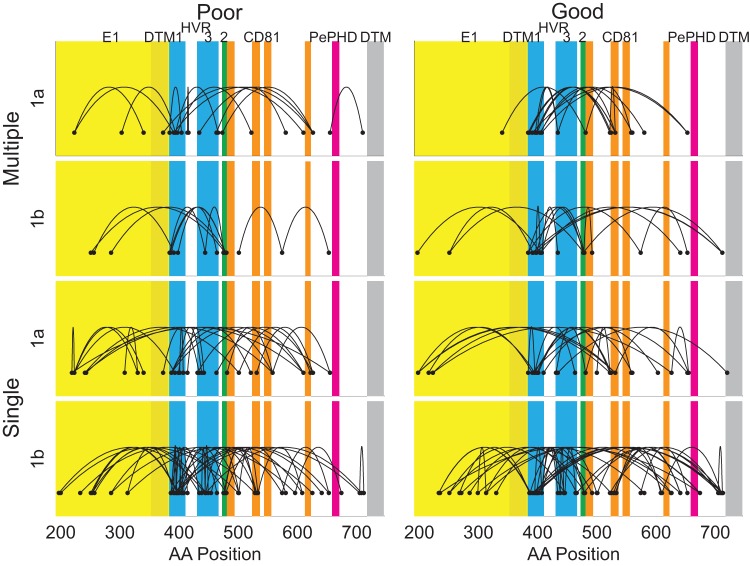
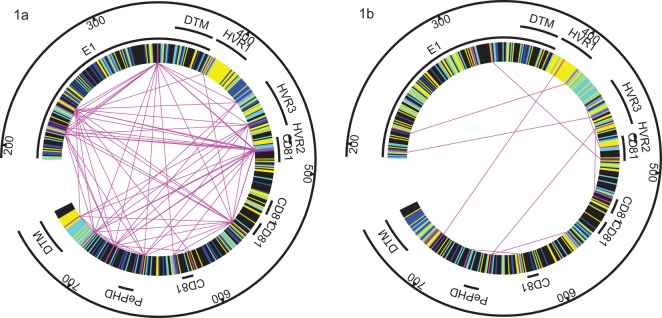
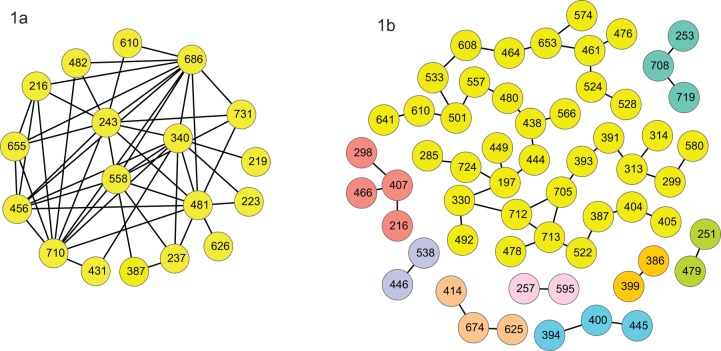
The poor response to the combined antiviral therapy of pegylated alfa-interferon and ribavarin for hepatitis C virus (HCV) infection may be linked to mutations in the viral envelope gene E1E2 (env), which can result in escape from the immune response and higher efficacy of viral entry. Mutations that result in failure of therapy most likely require compensatory mutations to achieve sufficient change in envelope structure and function. Compensatory mutations were investigated by determining positions in the E1E2 gene where amino acids (aa) covaried across groups of individuals. We assessed networks of covarying positions in E1E2 sequences that differentiated sustained virological response (SVR) from non-response (NR) in 43 genotype 1a (17 SVR), and 49 genotype 1b (25 SVR) chronically HCV-infected individuals. Binary integer programming over covariance networks was used to extract aa combinations that differed between response groups. Genotype 1a E1E2 sequences exhibited higher degrees of covariance and clustered into 3 main groups while 1b sequences exhibited no clustering. Between 5 and 9 aa pairs were required to separate SVR from NR in each genotype. aa in hypervariable region 1 were 6 times more likely than chance to occur in the optimal networks. The pair 531-626 (EI) appeared frequently in the optimal networks and was present in 6 of 9 NR in one of the 1a clusters. The most frequent pairs representing SVR were 431-481 (EE), 500-522 (QA) in 1a, and 407-434 (AQ) in 1b. Optimal networks based on covarying aa pairs in HCV envelope can indicate features that are associated with failure or success to antiviral therapy.
聚乙二醇干扰素和利巴韦林联合治疗丙型肝炎病毒 (HCV) 感染的反应不佳可能与病毒包膜基因 E1E2 (env) 的突变有关,这些突变可导致免疫逃逸和更高的病毒进入效率。导致治疗失败的突变很可能需要补偿性突变来实现包膜结构和功能的足够改变。通过确定 E1E2 基因中氨基酸 (aa) 在个体组之间共变的位置来研究补偿性突变。我们评估了 E1E2 序列中区分持续病毒学应答 (SVR) 和无应答 (NR) 的共变位置网络,这些网络区分了 43 名基因型 1a (17 名 SVR) 和 49 名基因型 1b (25 名 SVR) 慢性 HCV 感染个体。使用协方差网络上的二进制整数编程来提取在应答组之间存在差异的 aa 组合。基因型 1a 的 E1E2 序列表现出更高程度的协方差,并聚集成 3 个主要组,而 1b 序列则没有聚类。在每种基因型中,需要 5 到 9 对 aa 来区分 SVR 和 NR。高变区 1 中的 aa 发生在最佳网络中的可能性是随机的 6 倍。E1E2 中的 531-626 (EI) 对经常出现在最佳网络中,并且在 1a 中一个聚类的 9 个 NR 中有 6 个存在。代表 SVR 的最常见对是 431-481 (EE)、500-522 (QA) 在 1a 中,和 407-434 (AQ) 在 1b 中。基于 HCV 包膜共变 aa 对的最佳网络可以指示与抗病毒治疗失败或成功相关的特征。