Center for Systems Biology, Soochow University, Suzhou 215006, China.
J Transl Med. 2013 Jul 10;11:169. doi: 10.1186/1479-5876-11-169.
Clear cell renal cell carcinoma (ccRCC) represents the most invasive and common adult kidney neoplasm. Mounting evidence suggests that microRNAs (miRNAs) are important regulators of gene expression. But their function in tumourigenesis in this tumour type remains elusive. With the development of high throughput technologies such as microarrays and NGS, aberrant miRNA expression has been widely observed in ccRCC. Systematic and integrative analysis of multiple microRNA expression datasets may reveal potential mechanisms by which microRNAs contribute to ccRCC pathogenesis.
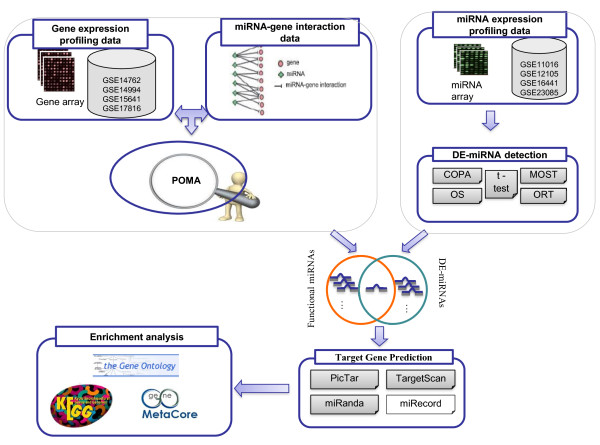
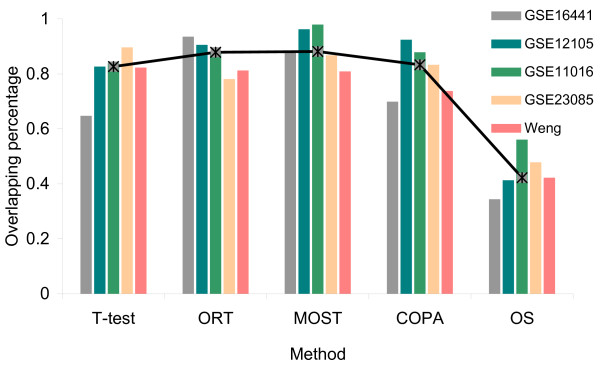
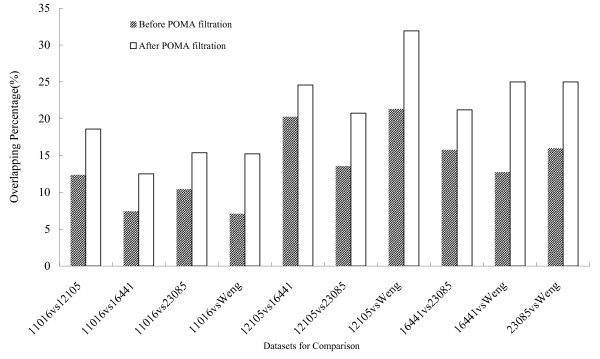
We collected 5 public microRNA expression datasets in ccRCC versus non-matching normal renal tissues from GEO database and published literatures. We analyzed these data sets with an integrated bioinformatics framework to identify expression signatures. The framework incorporates a novel statistic method for abnormal gene expression detection and an in-house developed predictor to assess the regulatory activity of microRNAs. We then mapped target genes of DE-miRNAs to different databases, such as GO, KEGG, GeneGo etc, for functional enrichment analysis.
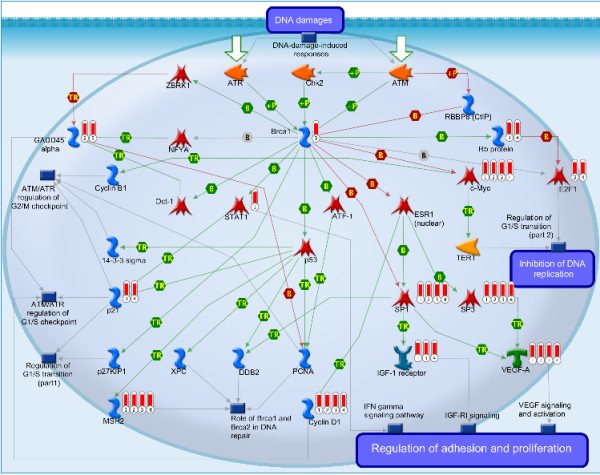
Using this framework we identified a consistent panel of eleven deregulated miRNAs shared by five independent datasets that can distinguish normal kidney tissues from ccRCC. After comparison with 3 RNA-seq based microRNA profiling studies, we found that our data correlated well with the results of next generation sequencing. We also discovered 14 novel molecular pathways that are likely to play a role in the tumourigenesis of ccRCC.
The integrative framework described in this paper greatly improves the inter-dataset consistency of microRNA expression signatures. Consensus expression profile should be identified at pathway or network level to address the heterogeneity of cancer. The DE-miRNA signature and novel pathways identified herein could provide potential biomarkers for ccRCC that await further validation.
透明细胞肾细胞癌(ccRCC)是最具侵袭性和最常见的成人肾肿瘤。越来越多的证据表明,microRNAs(miRNAs)是基因表达的重要调控因子。但它们在这种肿瘤类型中的肿瘤发生中的作用仍不清楚。随着高通量技术(如微阵列和 NGS)的发展,ccRCC 中已经广泛观察到异常的 miRNA 表达。对多个 miRNA 表达数据集进行系统和综合分析,可能揭示 miRNA 促进 ccRCC 发病机制的潜在机制。
我们从 GEO 数据库和已发表的文献中收集了 5 个公共的 ccRCC 与非匹配正常肾组织的 miRNA 表达数据集。我们使用集成的生物信息学框架来分析这些数据集,以识别表达特征。该框架结合了一种新的异常基因表达检测统计方法和一个内部开发的预测器,以评估 miRNA 的调控活性。然后,我们将 DE-miRNA 的靶基因映射到不同的数据库,如 GO、KEGG、GeneGo 等,进行功能富集分析。
使用该框架,我们确定了五个独立数据集之间共享的一组一致的 11 个失调 miRNA,可区分正常肾组织和 ccRCC。与 3 个基于 RNA-seq 的 miRNA 分析研究比较后,我们发现我们的数据与下一代测序的结果相关性良好。我们还发现了 14 个新的分子途径,这些途径可能在 ccRCC 的肿瘤发生中发挥作用。
本文描述的集成框架大大提高了 miRNA 表达特征在数据集之间的一致性。应该在途径或网络水平上确定一致的表达谱,以解决癌症的异质性。本文中确定的 DE-miRNA 特征和新途径可为 ccRCC 提供潜在的生物标志物,有待进一步验证。