Abou-Khalil Rana, Yang Frank, Mortreux Marie, Lieu Shirley, Yu Yan-Yiu, Wurmser Maud, Pereira Catia, Relaix Frédéric, Miclau Theodore, Marcucio Ralph S, Colnot Céline
INSERM U781, Université Paris Descartes-Sorbonne Paris Cité, Institut Imagine, Hôpital Necker Enfants Malades, Paris, France.
J Bone Miner Res. 2014 Feb;29(2):304-15. doi: 10.1002/jbmr.2038.
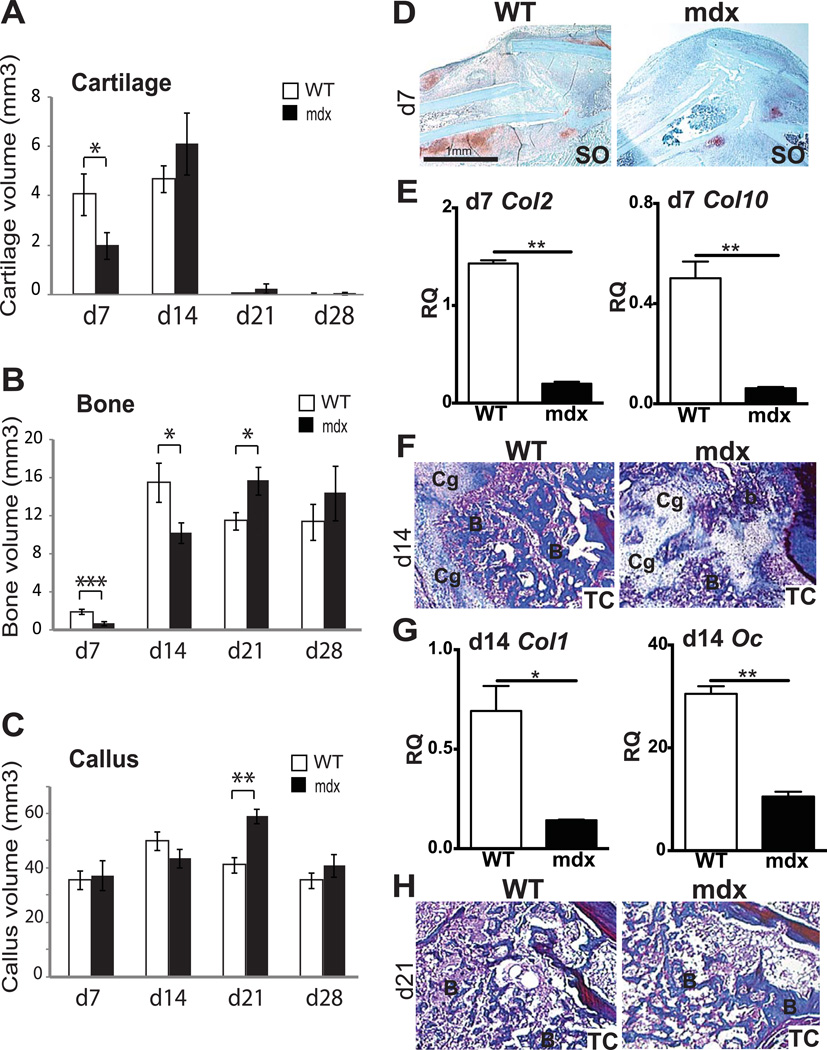
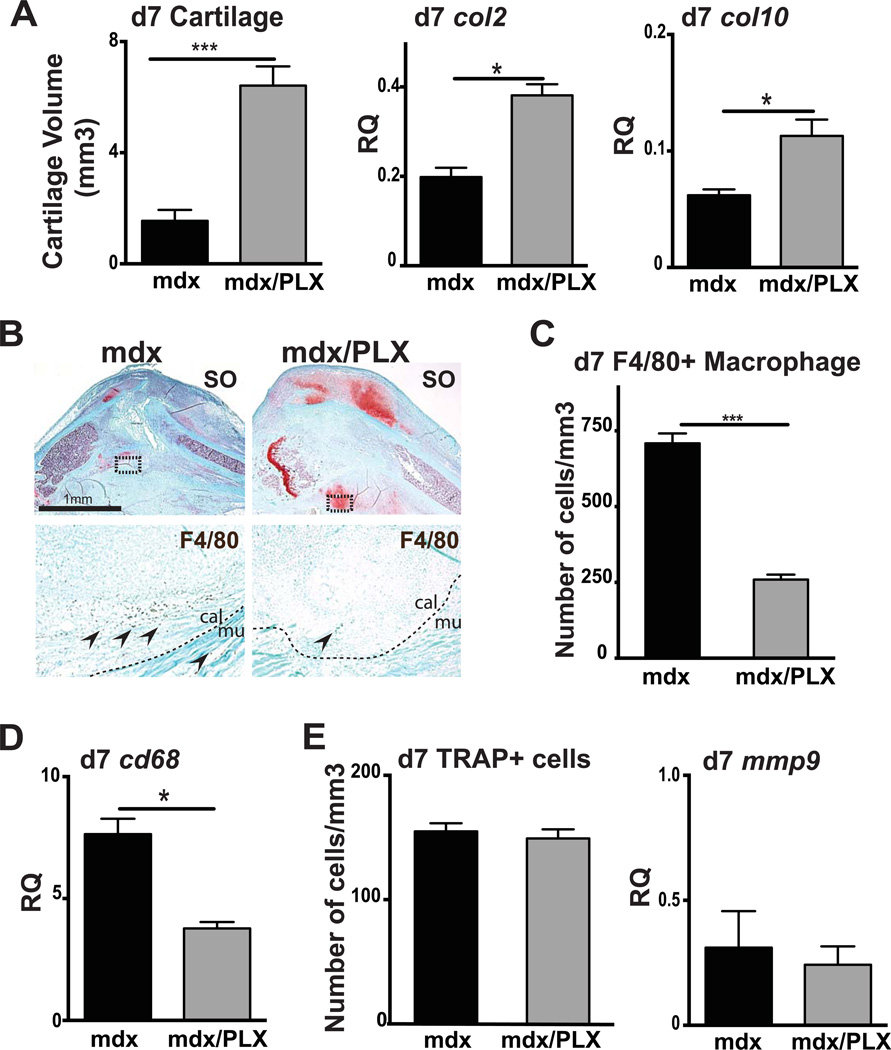
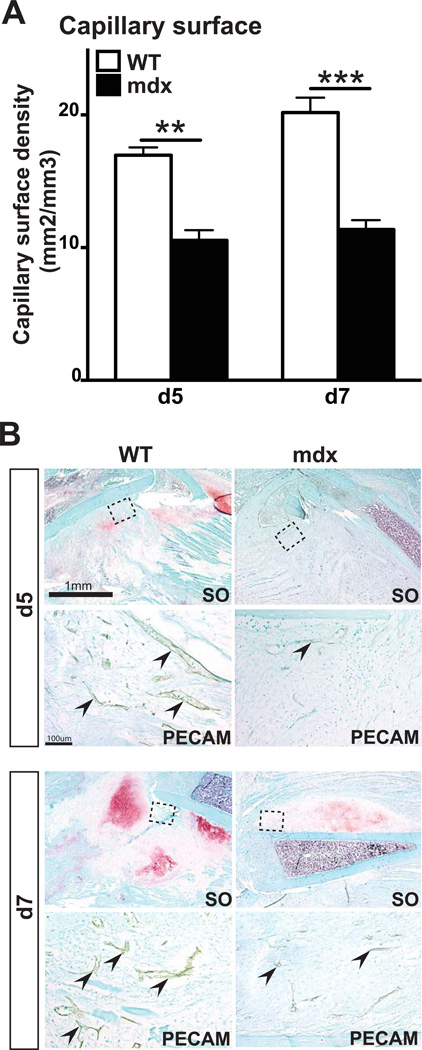
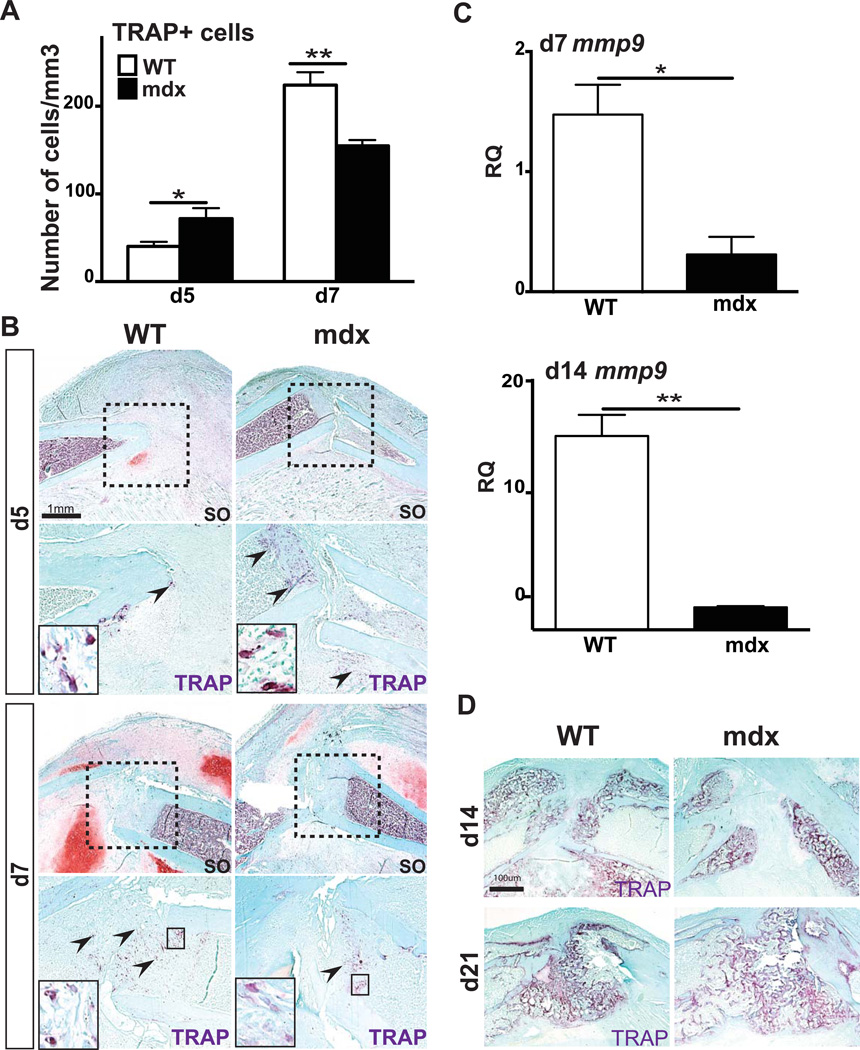
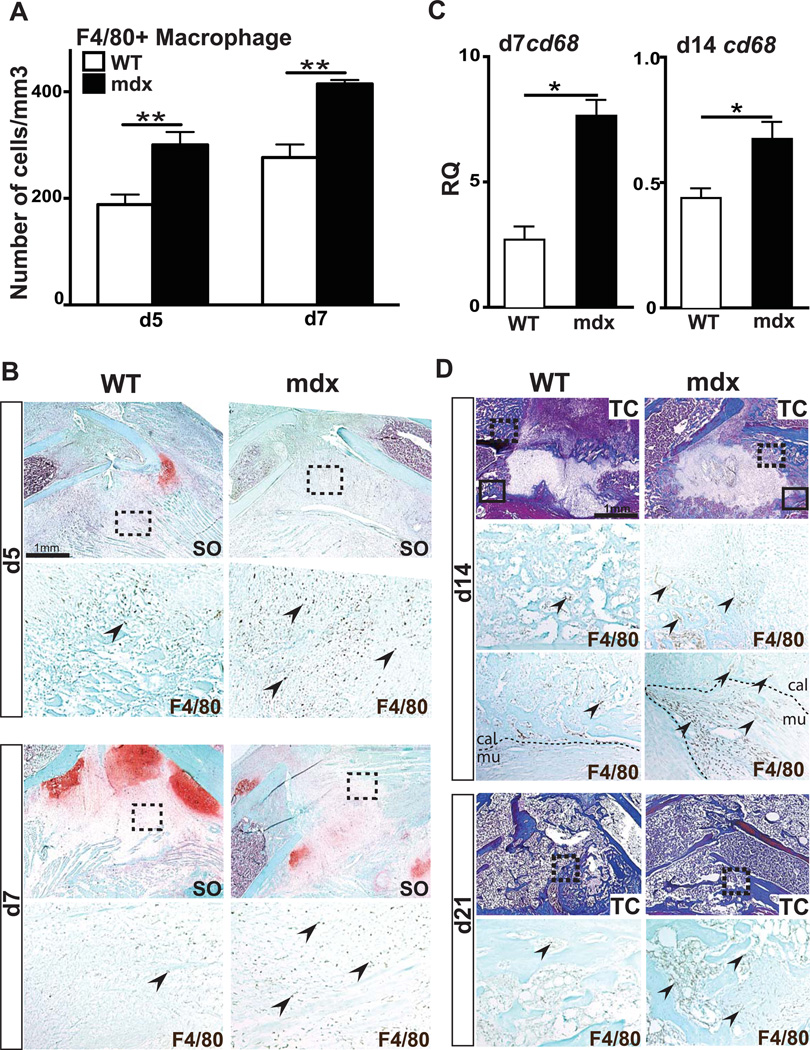
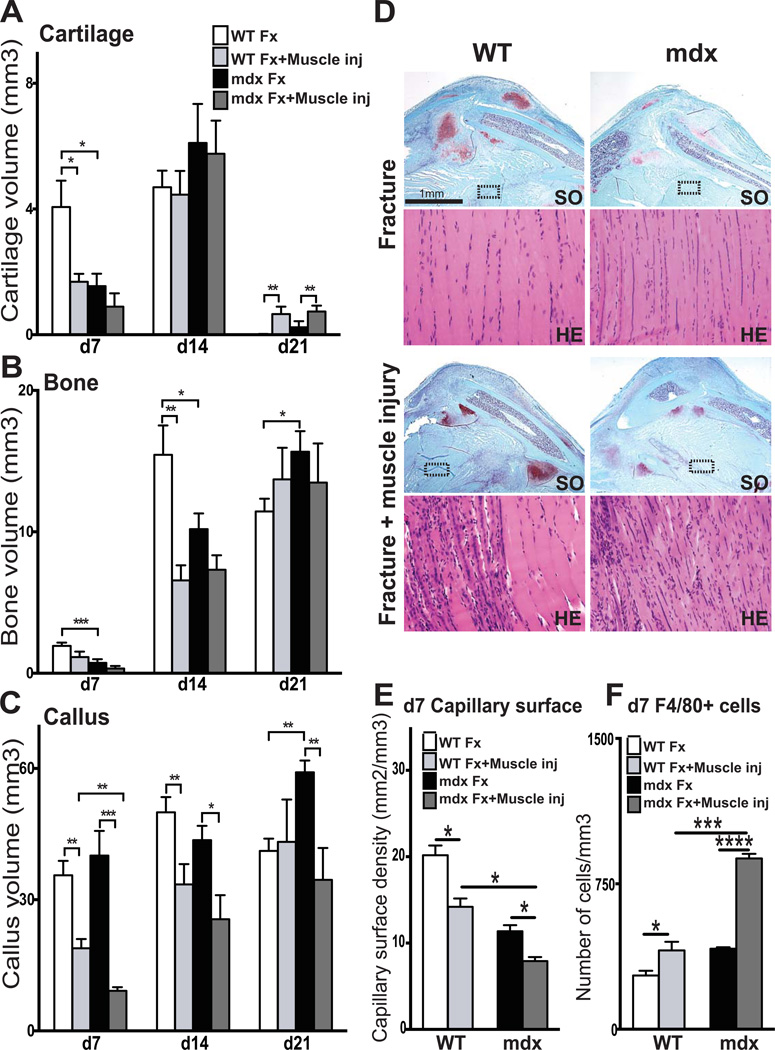
Duchenne muscular dystrophy (DMD) patients exhibit skeletal muscle weakness with continuous cycles of muscle fiber degeneration/regeneration, chronic inflammation, low bone mineral density, and increased risks of fracture. Fragility fractures and associated complications are considered as a consequence of the osteoporotic condition in these patients. Here, we aimed to establish the relationship between muscular dystrophy and fracture healing by assessing bone regeneration in mdx mice, a model of DMD with absence of osteoporosis. Our results illustrate that muscle defects in mdx mice impact the process of bone regeneration at various levels. In mdx fracture calluses, both cartilage and bone deposition were delayed followed by a delay in cartilage and bone remodeling. Vascularization of mdx fracture calluses was also decreased during the early stages of repair. Dystrophic muscles are known to contain elevated numbers of macrophages contributing to muscle degeneration. Accordingly, we observed increased macrophage recruitment in the mdx fracture calluses and abnormal macrophage accumulation throughout the process of bone regeneration. These changes in the inflammatory environment subsequently had an impact on the recruitment of osteoclasts and the remodeling phase of repair. Further damage to the mdx muscles, using a novel model of muscle trauma, amplified both the chronic inflammatory response and the delay in bone regeneration. In addition, PLX3397 treatment of mdx mice, a cFMS (colony stimulating factor receptor 1) inhibitor in monocytes, partially rescued the bone repair defect through increasing cartilage deposition and decreasing the number of macrophages. In conclusion, chronic inflammation in mdx mice contributes to the fracture healing delay and is associated with a decrease in angiogenesis and a transient delay in osteoclast recruitment. By revealing the role of dystrophic muscle in regulating the inflammatory response during bone repair, our results emphasize the implication of muscle in the normal bone repair process and may lead to improved treatment of fragility fractures in DMD patients.
杜氏肌营养不良症(DMD)患者表现出骨骼肌无力,伴有肌纤维变性/再生的持续循环、慢性炎症、低骨矿物质密度以及骨折风险增加。脆性骨折及相关并发症被认为是这些患者骨质疏松状态的结果。在此,我们旨在通过评估mdx小鼠(一种无骨质疏松的DMD模型)的骨再生来确定肌营养不良与骨折愈合之间的关系。我们的结果表明,mdx小鼠的肌肉缺陷在多个层面影响骨再生过程。在mdx骨折痂中,软骨和骨沉积均延迟,随后软骨和骨重塑也延迟。mdx骨折痂的血管化在修复早期也减少。已知营养不良的肌肉中巨噬细胞数量增加,这会导致肌肉变性。因此,我们观察到mdx骨折痂中巨噬细胞募集增加,并且在骨再生过程中巨噬细胞异常积聚。炎症环境的这些变化随后对破骨细胞的募集和修复的重塑阶段产生影响。使用一种新型肌肉创伤模型对mdx小鼠的肌肉造成进一步损伤,放大了慢性炎症反应和骨再生延迟。此外,用PLX3397(一种单核细胞中的cFMS(集落刺激因子受体1)抑制剂)治疗mdx小鼠,通过增加软骨沉积和减少巨噬细胞数量,部分挽救了骨修复缺陷。总之,mdx小鼠中的慢性炎症导致骨折愈合延迟,并与血管生成减少和破骨细胞募集的短暂延迟相关。通过揭示营养不良肌肉在调节骨修复过程中炎症反应的作用,我们的结果强调了肌肉在正常骨修复过程中的意义,并可能导致改善DMD患者脆性骨折的治疗。