Institut für Humangenetik, Universitätsklinikum Essen, Universität Duisburg-Essen, Essen, Germany.
Orphanet J Rare Dis. 2013 Jul 24;8:110. doi: 10.1186/1750-1172-8-110.
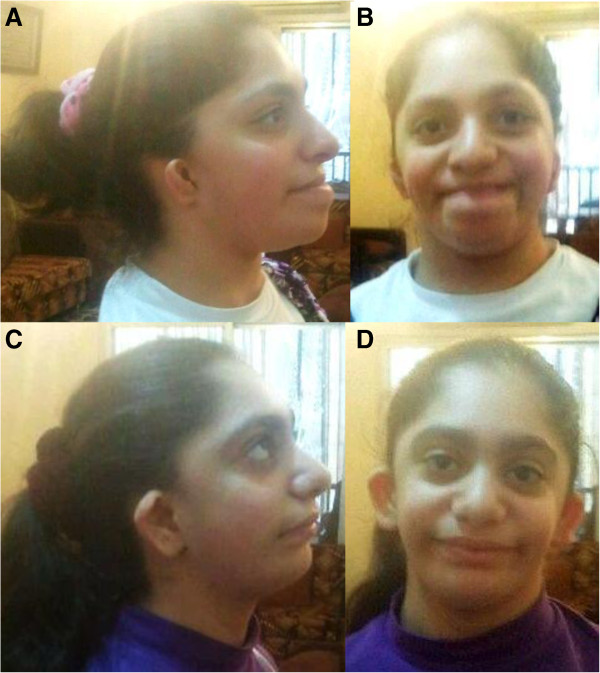
Mutations in EFTUD2 were proven to cause a very distinct mandibulofacial dysostosis type Guion-Almeida (MFDGA, OMIM #610536). Recently, gross deletions and mutations in EFTUD2 were determined to cause syndromic esophageal atresia (EA), as well. We set forth to find further conditions caused by mutations in the EFTUD2 gene (OMIM *603892).
We performed exome sequencing in two familial cases with clinical features overlapping with MFDGA and EA, but which were previously assumed to represent distinct entities, a syndrome with esophageal atresia, hypoplasia of zygomatic complex, microcephaly, cup-shaped ears, congenital heart defect, and intellectual disability in a mother and her two children [AJMG 143A(11):1135-1142, 2007] and a supposedly autosomal recessive oto-facial syndrome with midline malformations in two sisters [AJMG 132(4):398-401, 2005]. While the analysis of our exome data was in progress, a recent publication made EFTUD2 mutations highly likely in these families. This hypothesis could be confirmed with exome as well as with Sanger sequencing. Also, in three further sporadic patients, clinically overlapping to these two families, de novo mutations within EFTUD2 were identified by Sanger sequencing. Our clinical and molecular workup of the patients discloses a broad phenotypic spectrum, and describes for the first time an instance of germline mosaicism for an EFTUD2 mutation.
The clinical features of the eight patients described here further broaden the phenotypic spectrum caused by EFTUD2 mutations or deletions. We here show, that it not only includes mandibulofacial dysostosis type Guion-Almeida, which should be reclassified as an acrofacial dysostosis because of thumb anomalies (present in 12/35 or 34% of patients) and syndromic esophageal atresia [JMG 49(12). 737-746, 2012], but also the two new syndromes, namely oto-facial syndrome with midline malformations published by Mégarbané et al. [AJMG 132(4): 398-401, 2005] and the syndrome published by Wieczorek et al. [AJMG 143A(11): 1135-1142, 2007] The finding of mild phenotypic features in the mother of one family that could have been overlooked and the possibility of germline mosaicism in apparently healthy parents in the other family should be taken into account when counseling such families.
EFTUD2 基因突变被证实可导致一种非常独特的下颌面骨发育不全型 Guion-Almeida 综合征(MFDGA,OMIM#610536)。最近,EFTUD2 的大片段缺失和突变也被确定可导致综合征型食管闭锁(EA)。我们着手寻找由 EFTUD2 基因突变引起的其他疾病(OMIM*603892)。
我们对两例具有 MFDGA 和 EA 重叠临床表现的家系进行了外显子组测序,但之前认为它们代表不同的实体,一个是具有 EA、颧骨复合体发育不全、小头畸形、杯状耳、先天性心脏病和智力障碍的母亲及其两个孩子的综合征[AJMG 143A(11):1135-1142, 2007],另一个是两个姐妹的假定常染色体隐性耳面综合征伴中线畸形[AJMG 132(4):398-401, 2005]。在我们的外显子组数据分析进行的同时,最近的一篇文献使 EFTUD2 突变在这些家系中变得高度可能。这一假说可以通过外显子组和 Sanger 测序来证实。此外,在另外 3 例具有临床重叠表现的散发性患者中,通过 Sanger 测序鉴定了 EFTUD2 中的新生突变。我们对患者的临床和分子研究揭示了一个广泛的表型谱,并首次描述了 EFTUD2 突变的种系嵌合体。
本文描述的 8 例患者的临床特征进一步拓宽了由 EFTUD2 基因突变或缺失引起的表型谱。我们在此表明,它不仅包括下颌面骨发育不全型 Guion-Almeida,由于拇指异常(35 例患者中有 12 例,占 34%)和综合征型食管闭锁[JMG 49(12). 737-746, 2012],应重新归类为颅面骨发育不全,还包括 Mégarbané 等人发表的两种新综合征,即伴中线畸形的耳面综合征[AJMG 132(4): 398-401, 2005]和 Wieczorek 等人发表的综合征[AJMG 143A(11): 1135-1142, 2007]。一个家系中母亲的轻度表型特征可能被忽视,另一个家系中看似健康的父母中存在种系嵌合体,这些都应在为这些家系提供咨询时予以考虑。