School of Computer Science and Information Technology, Northeast Normal University, Changchun, People's Republic of China.
PLoS One. 2013 Jul 19;8(7):e69744. doi: 10.1371/journal.pone.0069744. Print 2013.
Although Transmembrane Proteins (TMPs) are highly important in various biological processes and pharmaceutical developments, general prediction of TMP structures is still far from satisfactory. Because TMPs have significantly different physicochemical properties from soluble proteins, current protein structure prediction tools for soluble proteins may not work well for TMPs. With the increasing number of experimental TMP structures available, template-based methods have the potential to become broadly applicable for TMP structure prediction. However, the current fold recognition methods for TMPs are not as well developed as they are for soluble proteins.
We developed a novel TMP Fold Recognition method, TMFR, to recognize TMP folds based on sequence-to-structure pairwise alignment. The method utilizes topology-based features in alignment together with sequence profile and solvent accessibility. It also incorporates a gap penalty that depends on predicted topology structure segments. Given the difference between α-helical transmembrane protein (αTMP) and β-strands transmembrane protein (βTMP), parameters of scoring functions are trained respectively for these two protein categories using 58 αTMPs and 17 βTMPs in a non-redundant training dataset.
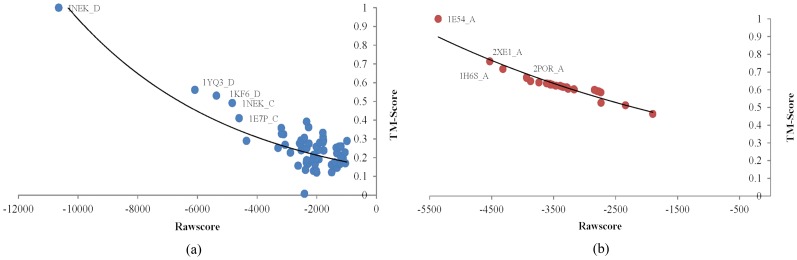
We compared our method with HHalign, a leading alignment tool using a non-redundant testing dataset including 72 αTMPs and 30 βTMPs. Our method achieved 10% and 9% better accuracies than HHalign in αTMPs and βTMPs, respectively. The raw score generated by TMFR is negatively correlated with the structure similarity between the target and the template, which indicates its effectiveness for fold recognition. The result demonstrates TMFR provides an effective TMP-specific fold recognition and alignment method.
尽管跨膜蛋白(TMP)在各种生物过程和药物开发中非常重要,但普遍预测 TMP 结构的方法仍远未令人满意。由于 TMP 的理化性质与可溶性蛋白有很大的不同,因此当前用于可溶性蛋白的蛋白质结构预测工具可能不适用于 TMP。随着实验 TMP 结构数量的增加,基于模板的方法有可能广泛适用于 TMP 结构预测。然而,目前用于 TMP 的折叠识别方法还不如可溶性蛋白那样发达。
我们开发了一种新的 TMP 折叠识别方法 TMFR,用于基于序列到结构的成对比对识别 TMP 折叠。该方法利用比对中的拓扑特征以及序列轮廓和溶剂可及性。它还结合了一种取决于预测拓扑结构段的间隙罚分。由于 α-螺旋跨膜蛋白(αTMP)和 β-折叠跨膜蛋白(βTMP)之间的差异,使用非冗余训练数据集(58 个 αTMP 和 17 个 βTMP)分别针对这两个蛋白质类别训练评分函数的参数。
我们使用非冗余测试数据集(包括 72 个 αTMP 和 30 个 βTMP)将我们的方法与 HHalign 进行了比较。在 αTMP 和 βTMP 中,我们的方法分别比 HHalign 高出 10%和 9%的准确性。TMFR 生成的原始分数与目标和模板之间的结构相似度呈负相关,这表明其对折叠识别的有效性。结果表明 TMFR 提供了一种有效的 TMP 特异性折叠识别和比对方法。