Pulmonary Disease Program, Vascular Biology Center, Georgia Health Sciences University, Augusta, Georgia, USA.
Compr Physiol. 2013 Jul;3(3):1011-34. doi: 10.1002/cphy.c120024.
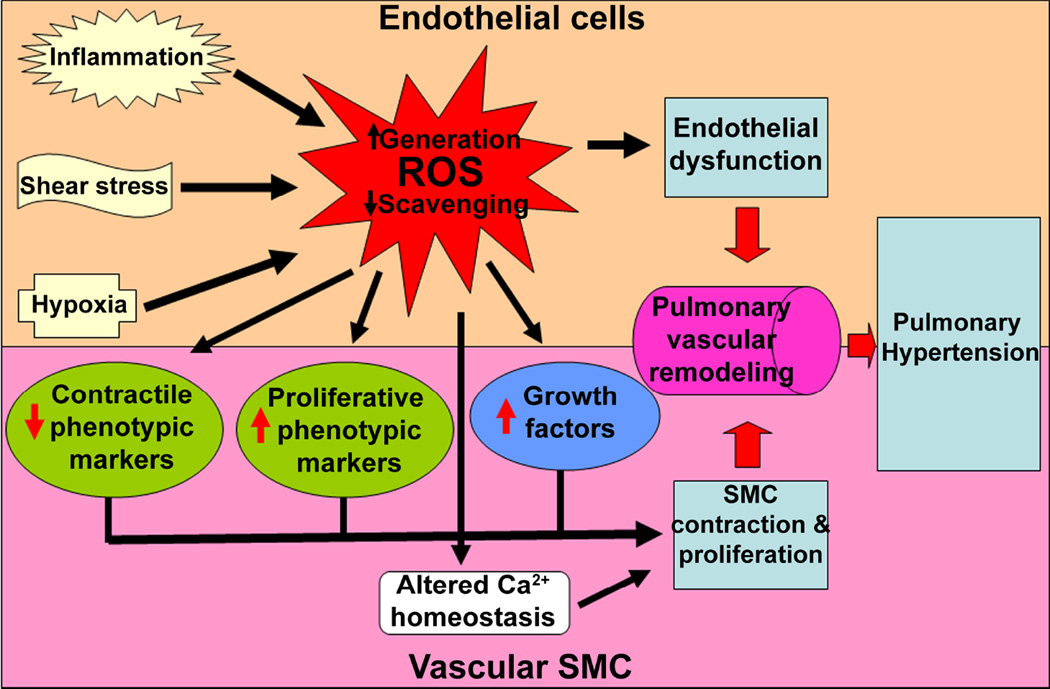
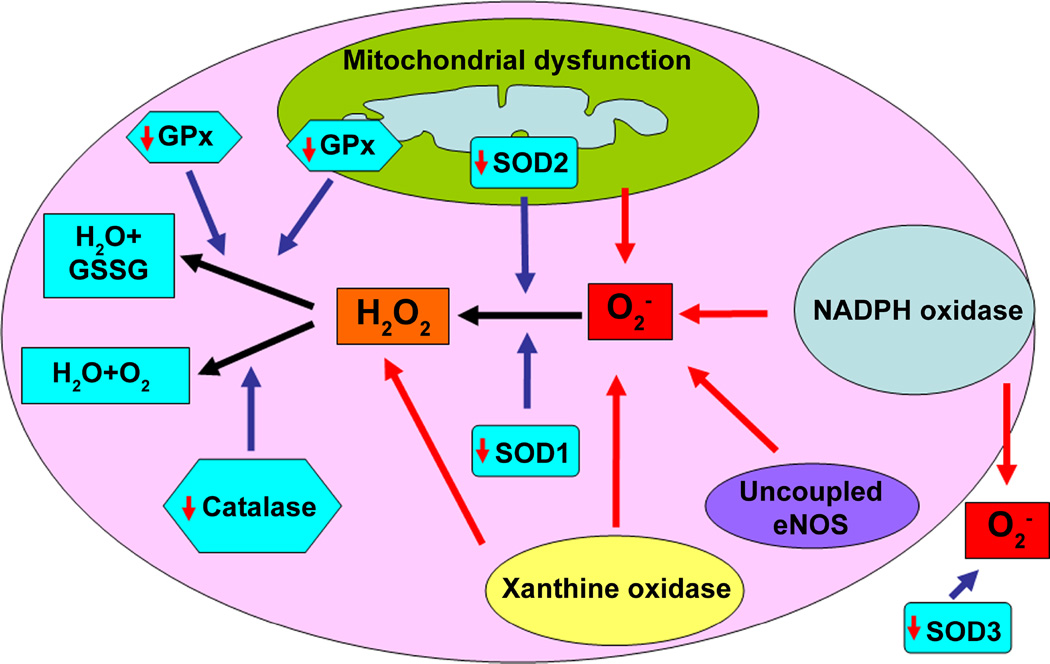
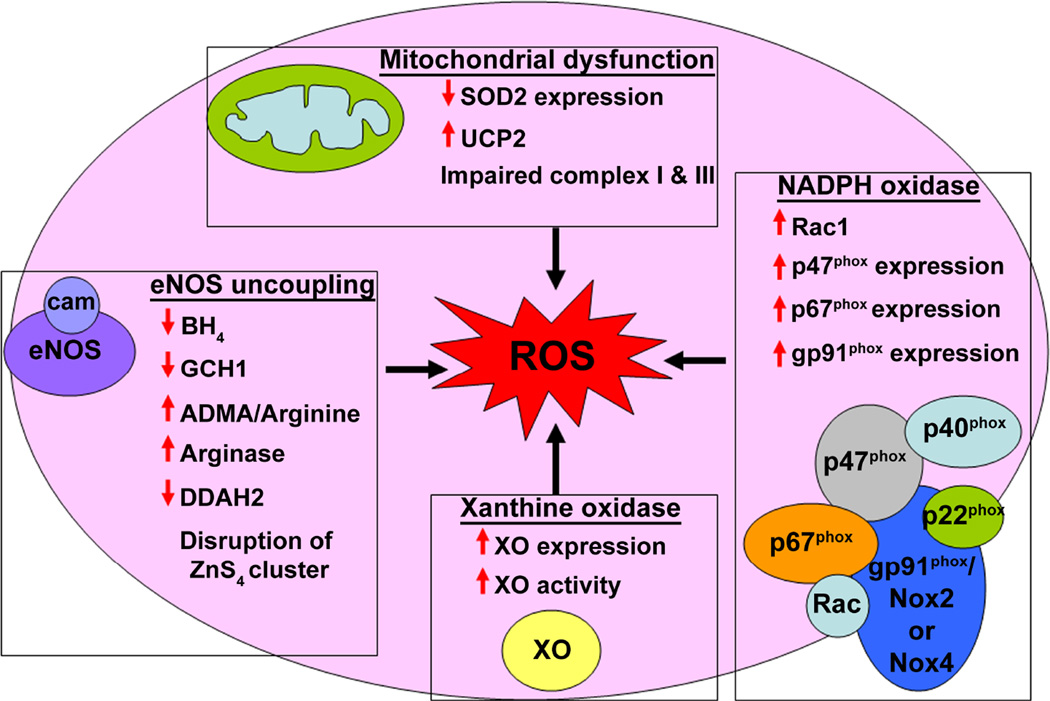
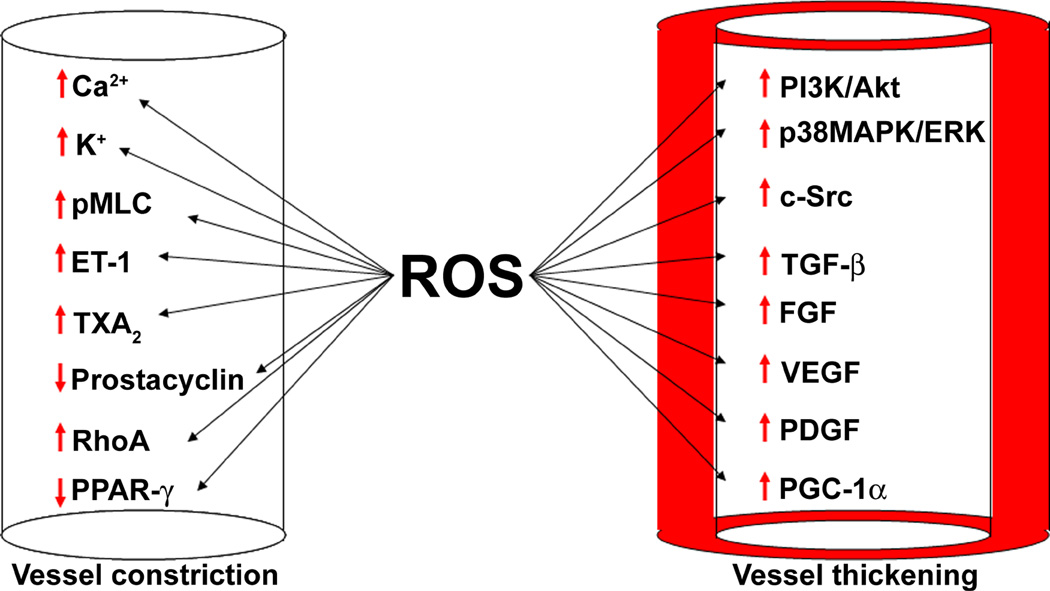
The pathogenesis of pulmonary hypertension is a complex multifactorial process that involves the remodeling of pulmonary arteries. This remodeling process encompasses concentric medial thickening of small arterioles, neomuscularization of previously nonmuscular capillary-like vessels, and structural wall changes in larger pulmonary arteries. The pulmonary arterial muscularization is characterized by vascular smooth muscle cell hyperplasia and hypertrophy. In addition, in uncontrolled pulmonary hypertension, the clonal expansion of apoptosis-resistant endothelial cells leads to the formation of plexiform lesions. Based upon a large number of studies in animal models, the three major stimuli that drive the vascular remodeling process are inflammation, shear stress, and hypoxia. Although, the precise mechanisms by which these stimuli impair pulmonary vascular function and structure are unknown, reactive oxygen species (ROS)-mediated oxidative damage appears to play an important role. ROS are highly reactive due to their unpaired valence shell electron. Oxidative damage occurs when the production of ROS exceeds the quenching capacity of the antioxidant mechanisms of the cell. ROS can be produced from complexes in the cell membrane (nicotinamide adenine dinucleotide phosphate-oxidase), cellular organelles (peroxisomes and mitochondria), and in the cytoplasm (xanthine oxidase). Furthermore, low levels of tetrahydrobiopterin (BH4) and L-arginine the rate limiting cofactor and substrate for endothelial nitric oxide synthase (eNOS), can cause the uncoupling of eNOS, resulting in decreased NO production and increased ROS production. This review will focus on the ROS generation systems, scavenger antioxidants, and oxidative stress associated alterations in vascular remodeling in pulmonary hypertension.
肺动脉高压的发病机制是一个复杂的多因素过程,涉及到肺血管的重塑。这个重塑过程包括小动脉的同心性中膜增厚、以前非肌性毛细血管样血管的新生肌化以及较大肺动脉的结构壁变化。肺血管的肌化表现为血管平滑肌细胞的增生和肥大。此外,在不受控制的肺动脉高压中,凋亡抵抗的内皮细胞的克隆扩增导致丛状病变的形成。基于大量的动物模型研究,驱动血管重塑过程的三个主要刺激因素是炎症、切应力和缺氧。尽管这些刺激因素损害肺血管功能和结构的确切机制尚不清楚,但活性氧(ROS)介导的氧化损伤似乎起着重要作用。由于其不成对的价壳电子,ROS 具有很高的反应性。当 ROS 的产生超过细胞抗氧化机制的淬灭能力时,就会发生氧化损伤。ROS 可以来自细胞膜中的复合物(烟酰胺腺嘌呤二核苷酸磷酸氧化酶)、细胞细胞器(过氧化物酶体和线粒体)和细胞质(黄嘌呤氧化酶)。此外,四氢生物蝶呤(BH4)和 L-精氨酸(内皮型一氧化氮合酶[eNOS]的限速辅助因子和底物)的水平降低,可导致 eNOS 的解偶联,从而导致 NO 生成减少和 ROS 生成增加。这篇综述将重点介绍 ROS 生成系统、清除抗氧化剂以及肺动脉高压中与血管重塑相关的氧化应激改变。