Division of Infectious Disease, Brigham & Women's Hospital, Boston, MA 02115, USA, Department of Microbiology and Immunobiology, Harvard Medical School, Boston, MA 02115, USA, Howard Hughes Medical Institute, Boston, MA 02115, USA, Department of Immunology and Infectious Diseases, Harvard School of Public Health, Boston, MA 02115, USA and Genome Sequencing and Analysis Program, Broad Institute, Cambridge, MA 02142, USA.
Nucleic Acids Res. 2013 Oct;41(19):9033-48. doi: 10.1093/nar/gkt654. Epub 2013 Jul 30.
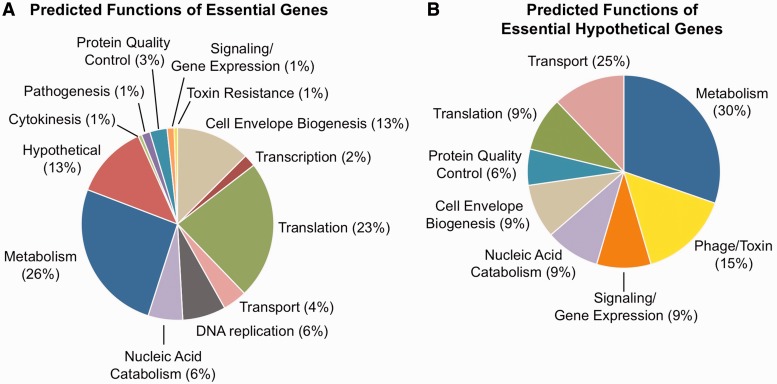
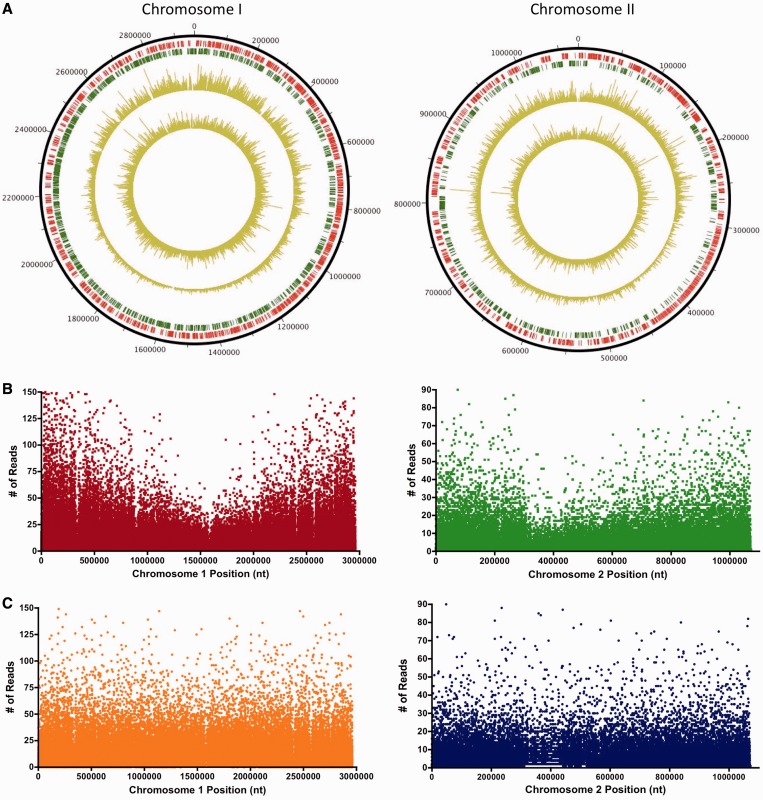
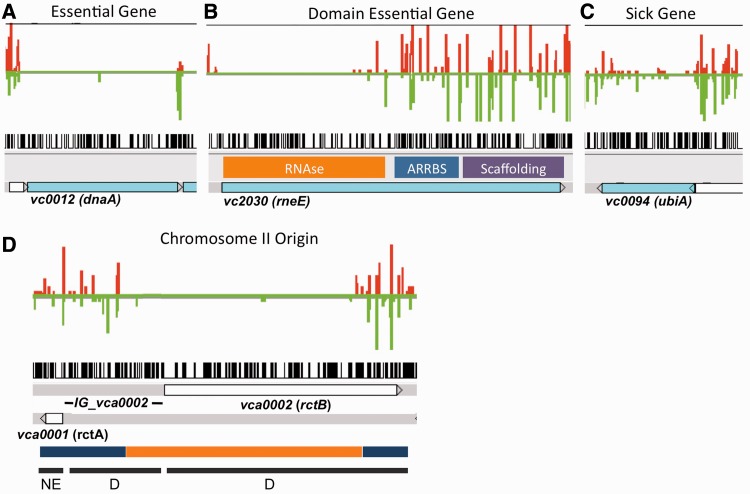
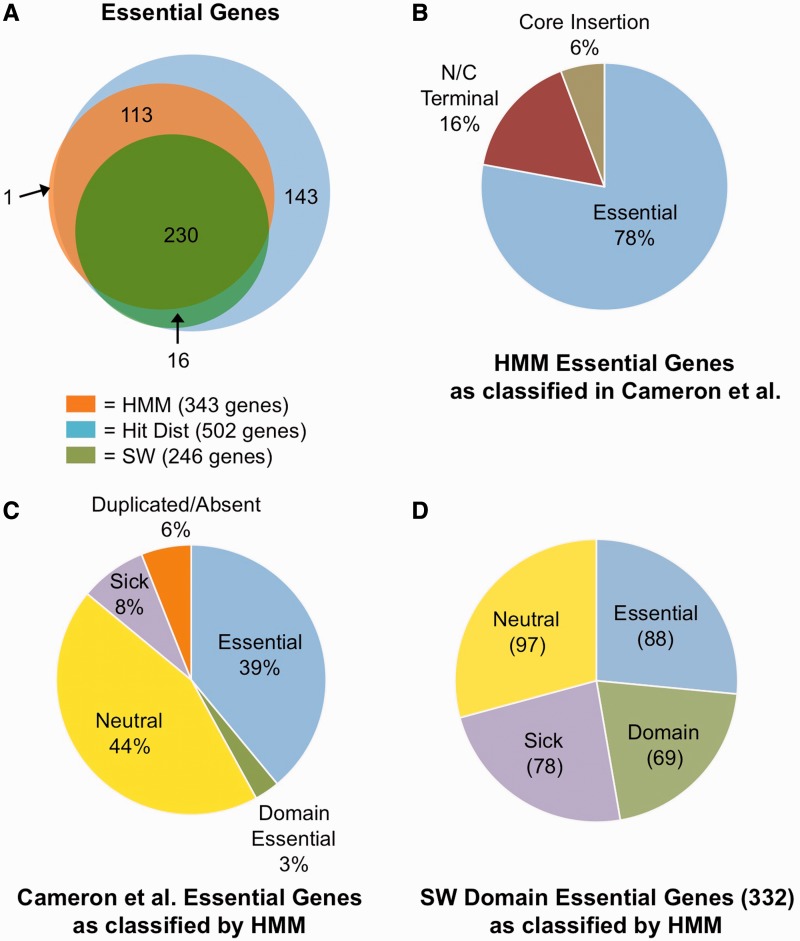
The coupling of high-density transposon mutagenesis to high-throughput DNA sequencing (transposon-insertion sequencing) enables simultaneous and genome-wide assessment of the contributions of individual loci to bacterial growth and survival. We have refined analysis of transposon-insertion sequencing data by normalizing for the effect of DNA replication on sequencing output and using a hidden Markov model (HMM)-based filter to exploit heretofore unappreciated information inherent in all transposon-insertion sequencing data sets. The HMM can smooth variations in read abundance and thereby reduce the effects of read noise, as well as permit fine scale mapping that is independent of genomic annotation and enable classification of loci into several functional categories (e.g. essential, domain essential or 'sick'). We generated a high-resolution map of genomic loci (encompassing both intra- and intergenic sequences) that are required or beneficial for in vitro growth of the cholera pathogen, Vibrio cholerae. This work uncovered new metabolic and physiologic requirements for V. cholerae survival, and by combining transposon-insertion sequencing and transcriptomic data sets, we also identified several novel noncoding RNA species that contribute to V. cholerae growth. Our findings suggest that HMM-based approaches will enhance extraction of biological meaning from transposon-insertion sequencing genomic data.
高密度转座子诱变与高通量 DNA 测序(转座子插入测序)相结合,能够同时对单个基因座对细菌生长和存活的贡献进行全基因组评估。我们通过对 DNA 复制对测序结果的影响进行归一化,并使用基于隐马尔可夫模型(HMM)的滤波器来利用所有转座子插入测序数据集固有的以前未被认识到的信息,从而改进了对转座子插入测序数据的分析。HMM 可以平滑读取丰度的变化,从而减少读取噪声的影响,还可以进行精细的图谱绘制,而无需基因组注释,并能够将基因座分类为几个功能类别(例如必需、域必需或“病态”)。我们生成了霍乱病原体霍乱弧菌体外生长所必需或有益的基因组基因座(包括内含子和外显子序列)的高分辨率图谱。这项工作揭示了霍乱弧菌生存的新代谢和生理需求,并且通过结合转座子插入测序和转录组数据集,我们还鉴定了几种有助于霍乱弧菌生长的新型非编码 RNA 种类。我们的研究结果表明,基于 HMM 的方法将增强从转座子插入测序基因组数据中提取生物学意义的能力。