Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA.
Nature. 2013 Aug 22;500(7463):477-81. doi: 10.1038/nature12433. Epub 2013 Aug 7.
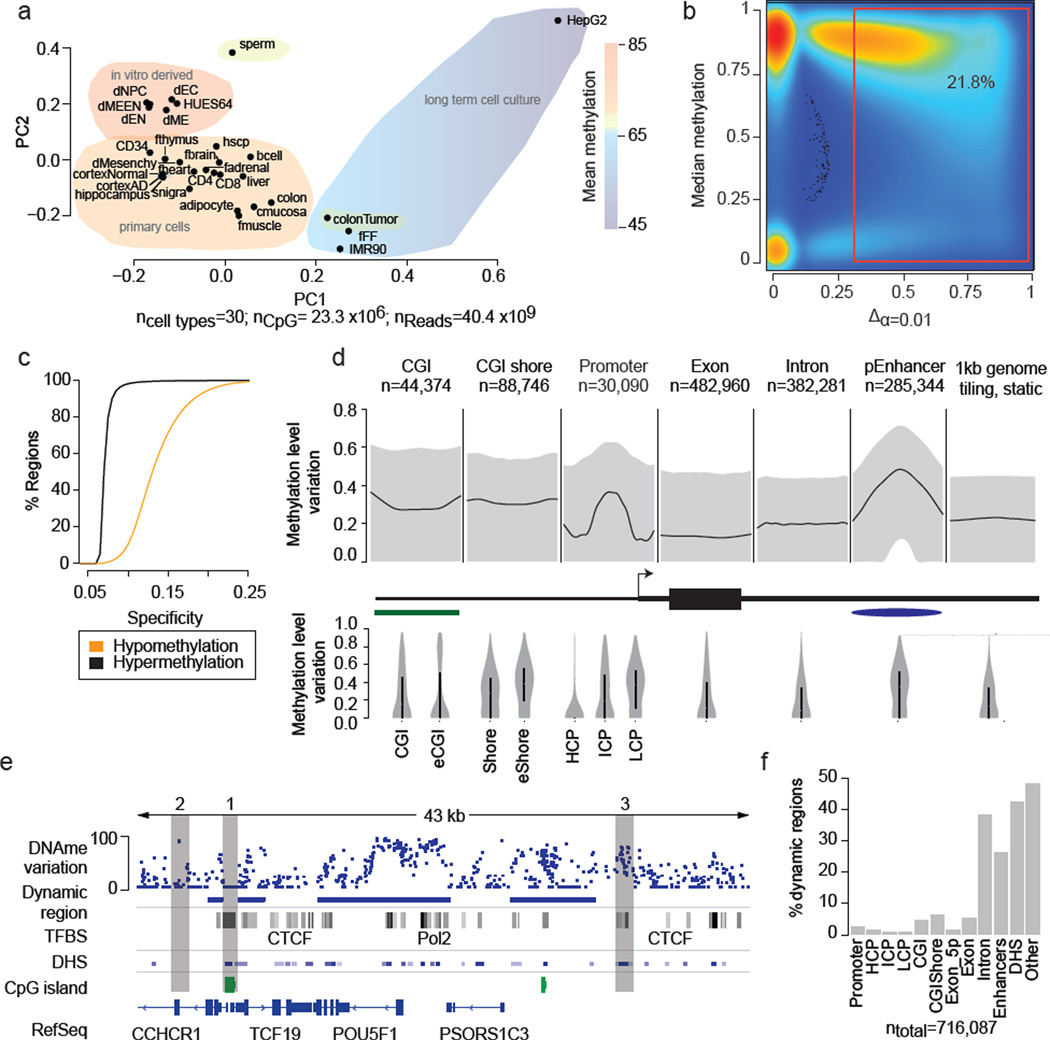
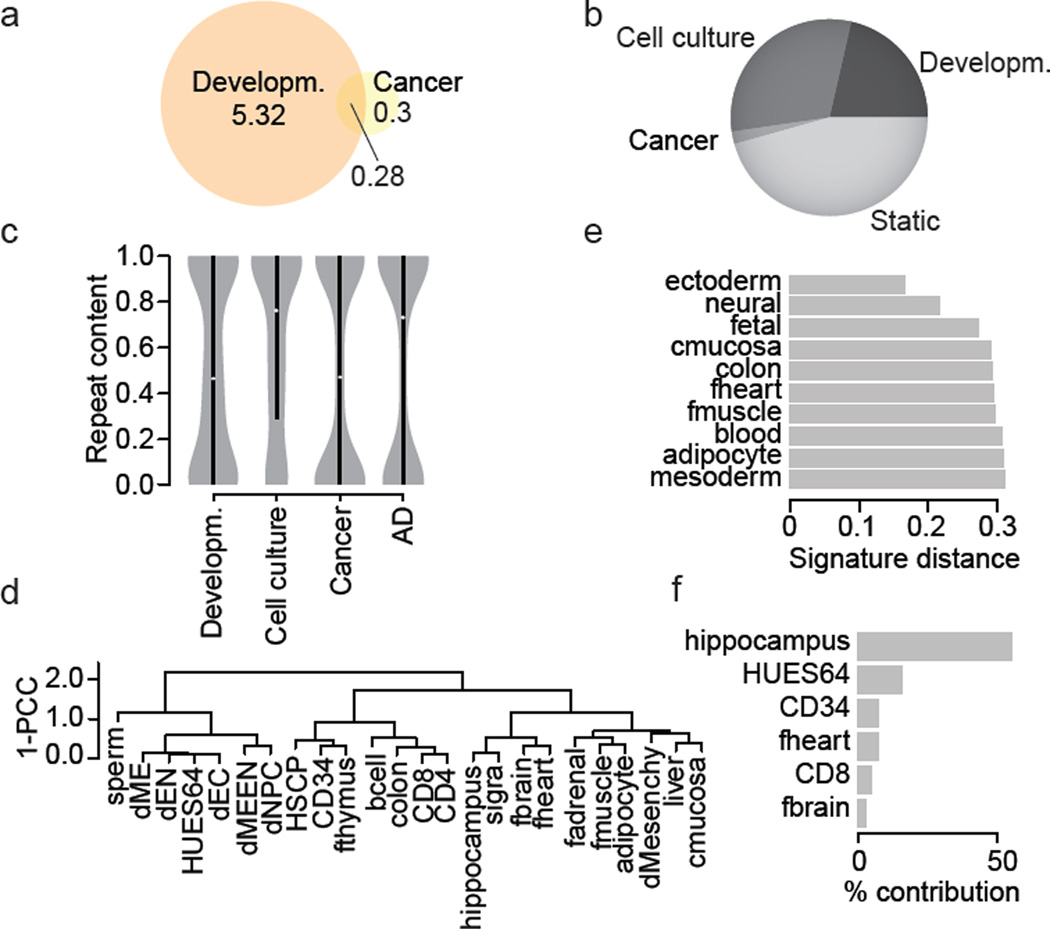
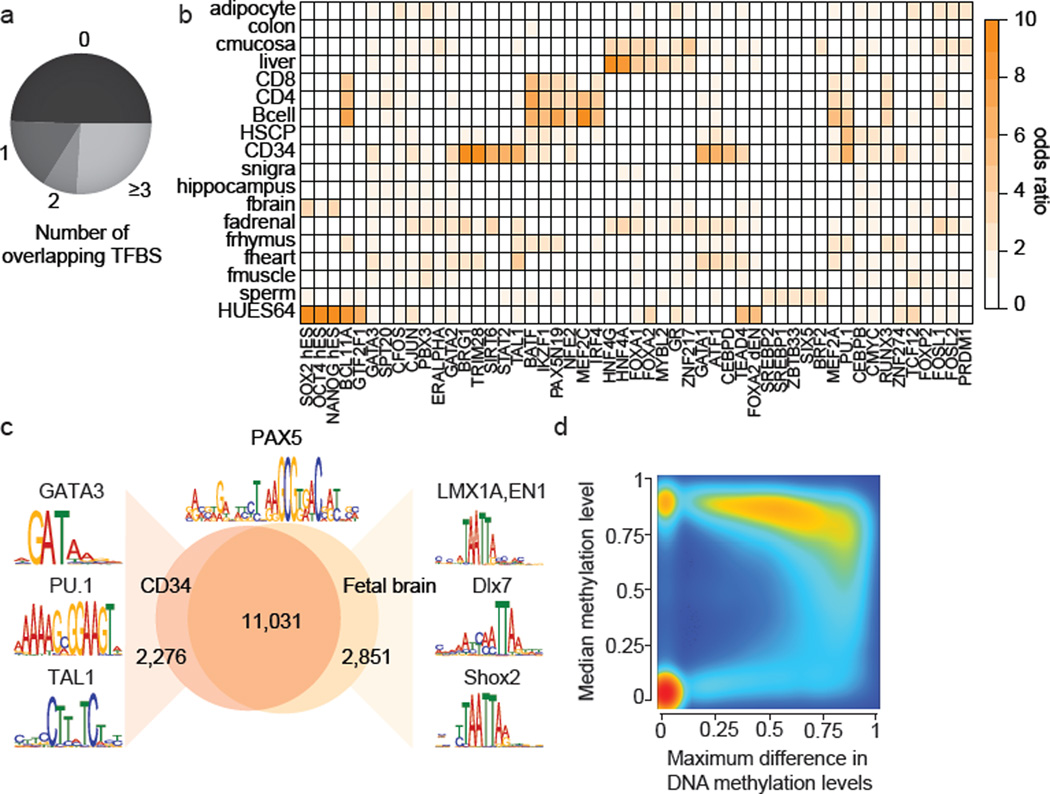
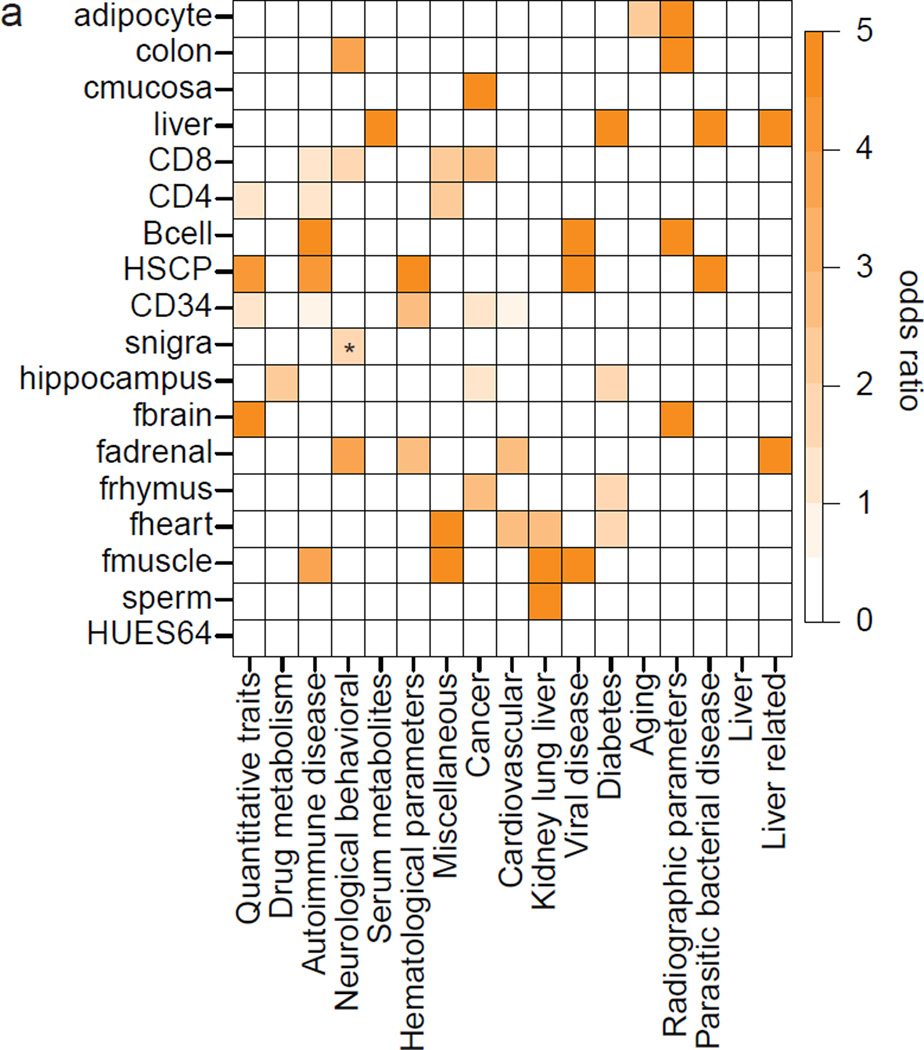
DNA methylation is a defining feature of mammalian cellular identity and is essential for normal development. Most cell types, except germ cells and pre-implantation embryos, display relatively stable DNA methylation patterns, with 70-80% of all CpGs being methylated. Despite recent advances, we still have a limited understanding of when, where and how many CpGs participate in genomic regulation. Here we report the in-depth analysis of 42 whole-genome bisulphite sequencing data sets across 30 diverse human cell and tissue types. We observe dynamic regulation for only 21.8% of autosomal CpGs within a normal developmental context, most of which are distal to transcription start sites. These dynamic CpGs co-localize with gene regulatory elements, particularly enhancers and transcription-factor-binding sites, which allow identification of key lineage-specific regulators. In addition, differentially methylated regions (DMRs) often contain single nucleotide polymorphisms associated with cell-type-related diseases as determined by genome-wide association studies. The results also highlight the general inefficiency of whole-genome bisulphite sequencing, as 70-80% of the sequencing reads across these data sets provided little or no relevant information about CpG methylation. To demonstrate further the utility of our DMR set, we use it to classify unknown samples and identify representative signature regions that recapitulate major DNA methylation dynamics. In summary, although in theory every CpG can change its methylation state, our results suggest that only a fraction does so as part of coordinated regulatory programs. Therefore, our selected DMRs can serve as a starting point to guide new, more effective reduced representation approaches to capture the most informative fraction of CpGs, as well as further pinpoint putative regulatory elements.
DNA 甲基化是哺乳动物细胞身份的决定性特征,对于正常发育至关重要。大多数细胞类型,除了生殖细胞和植入前胚胎外,都表现出相对稳定的 DNA 甲基化模式,所有 CpG 中有 70-80%被甲基化。尽管最近取得了进展,但我们对何时、何地以及有多少 CpG 参与基因组调控仍然知之甚少。在这里,我们报告了对 30 个人类细胞和组织类型的 42 个全基因组亚硫酸氢盐测序数据集的深入分析。我们观察到在正常发育背景下,仅有 21.8%的常染色体 CpG 发生动态调控,其中大多数位于转录起始位点的远端。这些动态 CpG 与基因调控元件,特别是增强子和转录因子结合位点共定位,这使得能够鉴定出关键的谱系特异性调节剂。此外,差异甲基化区域(DMRs)通常包含与细胞类型相关疾病相关的单核苷酸多态性,这是通过全基因组关联研究确定的。结果还突出了全基因组亚硫酸氢盐测序的普遍低效性,因为在这些数据集的 70-80%的测序reads 几乎没有或没有提供关于 CpG 甲基化的相关信息。为了进一步展示我们的 DMR 集的实用性,我们使用它对未知样本进行分类,并确定代表性的特征区域,这些区域可以重现主要的 DNA 甲基化动态。总之,尽管从理论上讲,每个 CpG 都可以改变其甲基化状态,但我们的结果表明,只有一部分 CpG 会作为协调的调控程序的一部分发生这种变化。因此,我们选择的 DMR 可以作为一个起点,指导新的、更有效的代表性降低方法,以捕获最具信息量的 CpG 部分,并进一步确定潜在的调控元件。