Novartis Institute for Tropical Diseases, Singapore, Singapore.
PLoS One. 2013 Jul 31;8(7):e69191. doi: 10.1371/journal.pone.0069191. Print 2013.
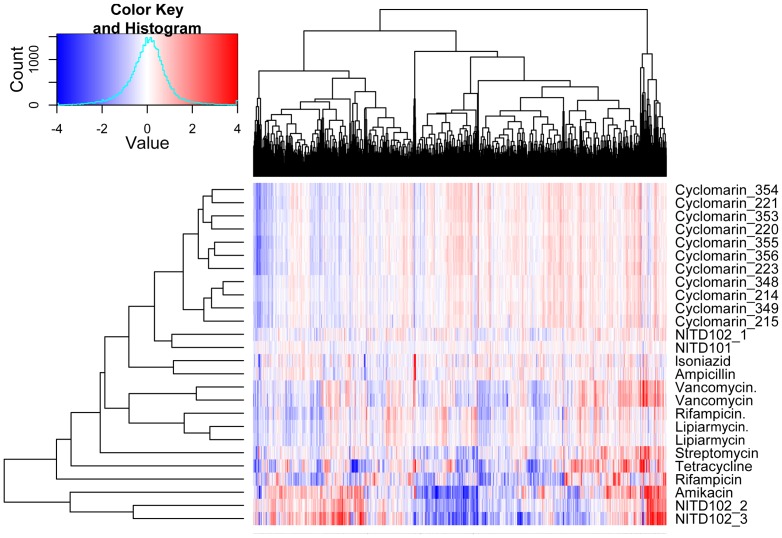
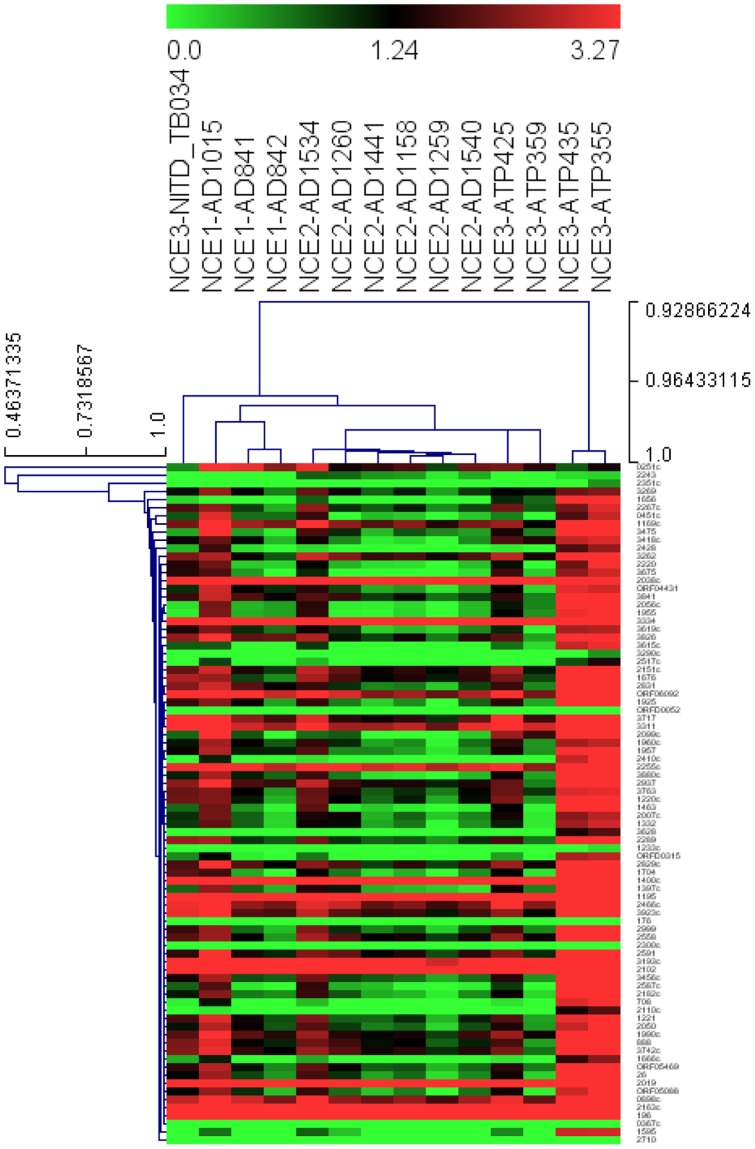
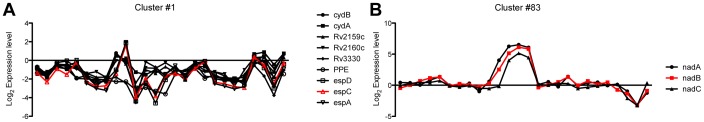
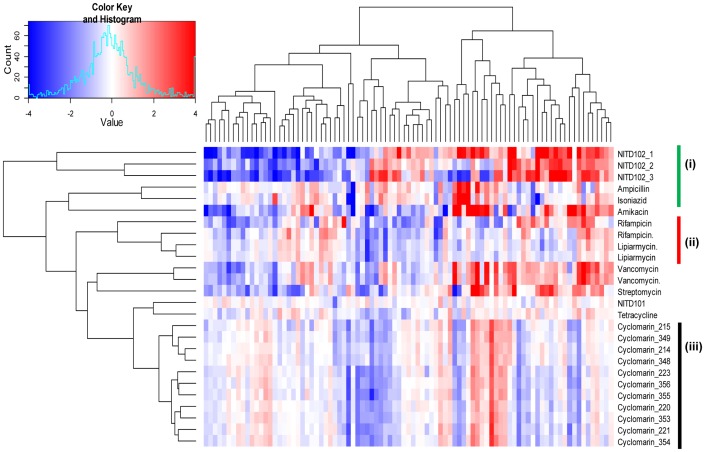
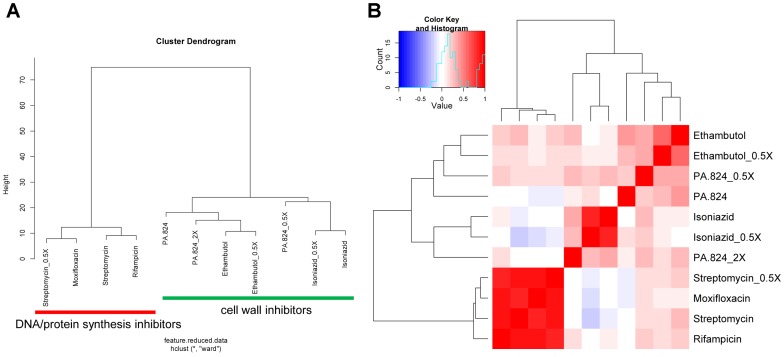
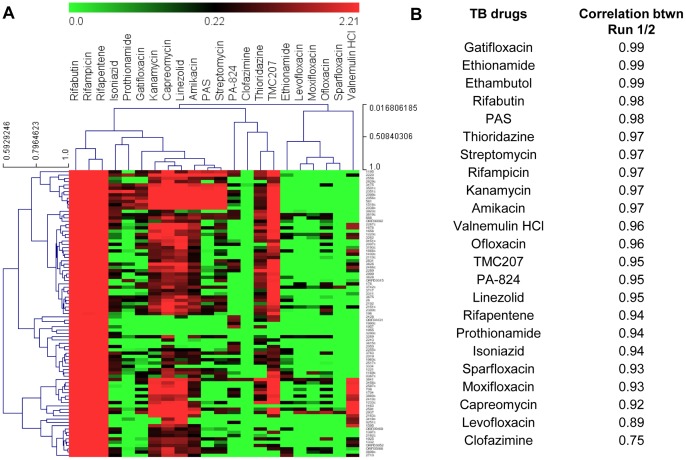
Most candidate anti-bacterials are identified on the basis of their whole cell anti-bacterial activity. A critical bottleneck in the early discovery of novel anti-bacterials is tracking the structure activity relationship (SAR) of the novel compounds synthesized during the hit to lead and lead optimization stage. It is often very difficult for medicinal chemists to visualize if the novel compounds synthesized for understanding SAR of a particular scaffold have similar molecular mechanism of action (MoA) as that of the initial hit. The elucidation of the molecular MoA of bioactive inhibitors is critical. Here, a new strategy and routine assay for MoA de-convolution, using a microfluidic platform for transcriptional profiling of bacterial response to inhibitors with whole cell activity has been presented. First a reference transcriptome compendium of Mycobacterial response to various clinical and investigational drugs was built. Using feature reduction, it was demonstrated that subsets of biomarker genes representative of the whole genome are sufficient for MoA classification and deconvolution in a medium-throughput microfluidic format ultimately leading to a cost effective and rapid tool for routine antibacterial drug-discovery programs.
大多数候选抗菌药物是根据其全细胞抗菌活性来确定的。在新型抗菌药物的早期发现中,一个关键的瓶颈是跟踪在命中到先导化合物和先导化合物优化阶段合成的新型化合物的结构活性关系(SAR)。对于药物化学家来说,要直观地了解为了理解特定支架的 SAR 而合成的新型化合物是否与初始命中具有相似的作用机制(MoA),往往非常困难。阐明生物活性抑制剂的分子 MoA 至关重要。在这里,提出了一种使用微流控平台对具有全细胞活性的抑制剂进行细菌转录谱分析的用于 MoA 去卷积的新策略和常规测定方法。首先,构建了结核分枝杆菌对各种临床和研究药物反应的参考转录组概要。通过特征减少,证明代表整个基因组的生物标志物基因子集足以用于中高通量微流控格式中的 MoA 分类和去卷积,最终为常规抗菌药物发现计划提供了一种具有成本效益且快速的工具。