Department of Neurosurgery, University Hospital Tuebingen, Hoppe-Seyler-Str. 3, 72076, Tuebingen, Baden-Wuerttemberg, Germany.
BMC Neurol. 2013 Aug 15;13:107. doi: 10.1186/1471-2377-13-107.
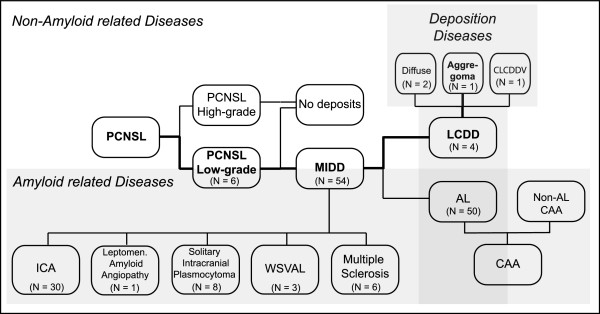
This work aims to add evidence and provide an update on the classification and diagnosis of monoclonal immunoglobulin deposition disease (MIDD) and primary central nervous system low-grade lymphomas. MIDD is characterized by the deposition of light and heavy chain proteins. Depending on the spatial arrangement of the secreted proteins, light chain-derived amyloidosis (AL) can be distinguished from non-amyloid light chain deposition disease (LCDD). We present a case of an extremely rare tumoral presentation of LCDD (aggregoma) and review the 3 previously published LCDD cases and discuss their presentation with respect to AL.
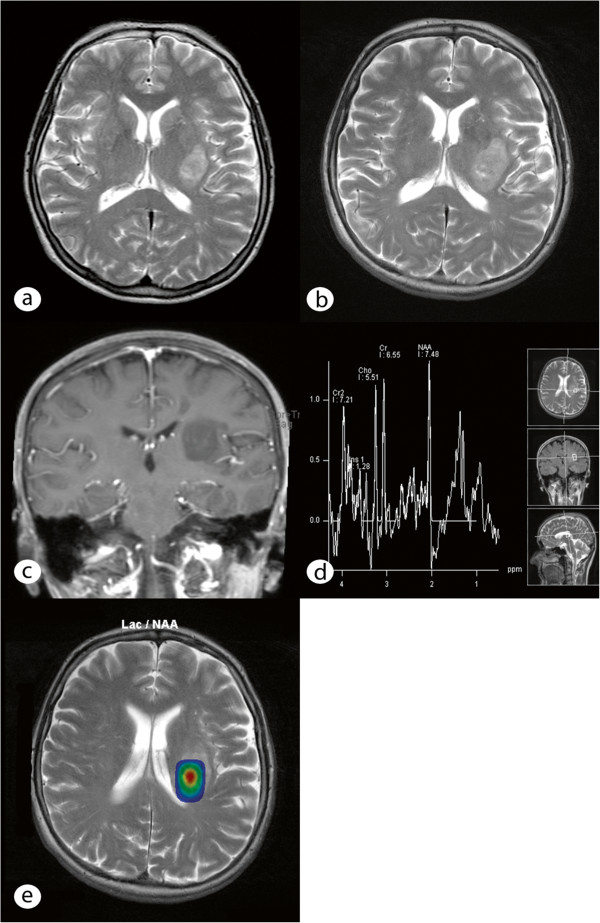
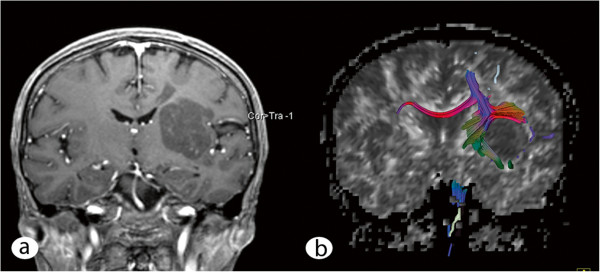
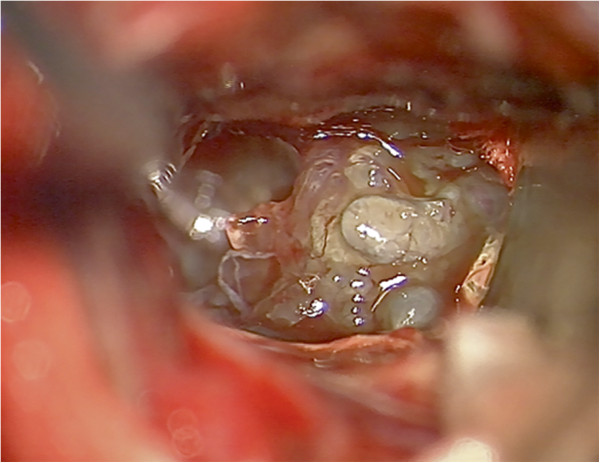
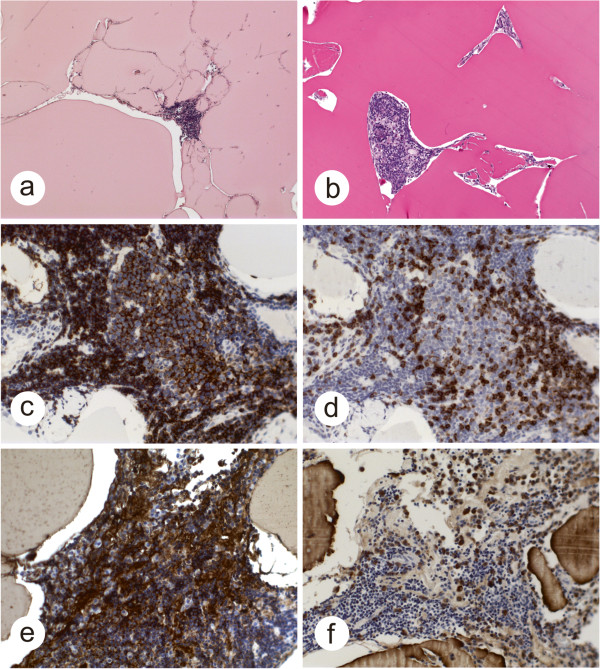
A 61-year-old woman presented with a 3½-year history of neurologic symptoms due to a progressive white matter lesion of the left subcortical parieto-insular lobe and basal ganglia. 2 former stereotactic biopsies conducted at different hospitals revealed no evidence of malignancy or inflammation; thus, no therapy had been initiated. After performing physiological and functional magnetic resonance imaging (MRI), the tumor was removed under intraoperative monitoring at our department. Histological analysis revealed large amorphous deposits and small islands of lymphoid cells.
LCCD is a very rare and obscure manifestation of primary central nervous system low-grade lymphomas that can be easily misdiagnosed by stereotactic biopsy sampling. If stereotactic biopsy does not reveal a definite result, a "wait-and-see" strategy can delay possible therapy for this disease. The impact of surgical removal, radiotherapy and chemotherapy in LCDD obviously remains controversial because of the low number of relevant cases.
本研究旨在为单克隆免疫球蛋白沉积病(MIDD)和原发性中枢神经系统低度淋巴瘤的分类和诊断提供新的证据和更新。MIDD 的特征是轻链和重链蛋白的沉积。根据分泌蛋白的空间排列,可将轻链衍生的淀粉样变性(AL)与非淀粉样轻链沉积病(LCDD)区分开来。我们报告了一例极为罕见的 LCDD(聚集瘤)的肿瘤表现,并复习了之前发表的 3 例 LCDD 病例,并就其与 AL 的表现进行了讨论。
一名 61 岁女性,因左皮质下顶叶岛叶和基底节的进行性白质病变出现 3 年半的神经症状。前两家医院进行的 2 次立体定向活检未发现恶性或炎症的证据,因此未进行治疗。在进行生理和功能磁共振成像(MRI)后,我们科室在术中监测下切除了肿瘤。组织学分析显示有大量无定形沉积物和小淋巴细胞岛。
LCDD 是原发性中枢神经系统低度淋巴瘤非常罕见和隐匿的表现,通过立体定向活检取样很容易误诊。如果立体定向活检没有得出明确的结果,可以采取“观望”策略来延迟这种疾病的可能治疗。由于相关病例数量较少,手术切除、放疗和化疗对 LCDD 的影响显然仍存在争议。