Department of Biostatistics and Epidemiology, University of Pennsylvania Perelman School of Medicine Philadelphia, PA, USA.
Front Genet. 2013 Aug 16;4:157. doi: 10.3389/fgene.2013.00157. eCollection 2013.
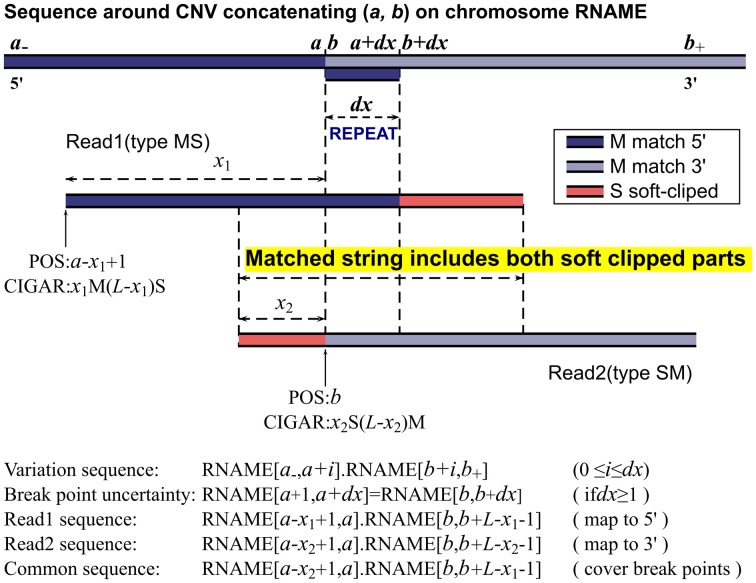
Copy number variations (CNVs) are associated with many complex diseases. Next generation sequencing data enable one to identify precise CNV breakpoints to better under the underlying molecular mechanisms and to design more efficient assays. Using the CIGAR strings of the reads, we develop a method that can identify the exact CNV breakpoints, and in cases when the breakpoints are in a repeated region, the method reports a range where the breakpoints can slide. Our method identifies the breakpoints of a CNV using both the positions and CIGAR strings of the reads that cover breakpoints of a CNV. A read with a long soft clipped part (denoted as S in CIGAR) at its 3'(right) end can be used to identify the 5'(left)-side of the breakpoints, and a read with a long S part at the 5' end can be used to identify the breakpoint at the 3'-side. To ensure both types of reads cover the same CNV, we require the overlapped common string to include both of the soft clipped parts. When a CNV starts and ends in the same repeated regions, its breakpoints are not unique, in which case our method reports the left most positions for the breakpoints and a range within which the breakpoints can be incremented without changing the variant sequence. We have implemented the methods in a C++ package intended for the current Illumina Miseq and Hiseq platforms for both whole genome and exon-sequencing. Our simulation studies have shown that our method compares favorably with other similar methods in terms of true discovery rate, false positive rate and breakpoint accuracy. Our results from a real application have shown that the detected CNVs are consistent with zygosity and read depth information. The software package is available at http://statgene.med.upenn.edu/softprog.html.
拷贝数变异 (CNVs) 与许多复杂疾病有关。下一代测序数据可用于识别精确的 CNV 断点,以更好地了解潜在的分子机制,并设计更有效的检测方法。我们利用测序reads 的 CIGAR 字符串开发了一种方法,可以识别确切的 CNV 断点,并且在断点位于重复区域的情况下,该方法报告了断点可以滑动的范围。我们的方法使用覆盖 CNV 断点的 reads 的位置和 CIGAR 字符串来识别 CNV 的断点。在其 3'(右)末端具有长软剪接部分(在 CIGAR 中表示为 S)的 read 可用于识别断点的 5'(左)侧,并且在 5' 末端具有长 S 部分的 read 可用于识别 3'侧的断点。为了确保这两种类型的 reads 都覆盖相同的 CNV,我们要求重叠的公共字符串包含两个软剪接部分。当 CNV 在相同的重复区域中开始和结束时,其断点不是唯一的,在这种情况下,我们的方法报告断点的最左侧位置以及可以在不改变变异序列的情况下递增断点的范围内。我们已经在一个 C++ 包中实现了这些方法,该包针对当前的 Illumina Miseq 和 Hiseq 平台,用于全基因组和外显子测序。我们的模拟研究表明,在真发现率、假阳性率和断点准确性方面,我们的方法与其他类似方法相比具有优势。我们在真实应用中的结果表明,检测到的 CNVs 与同卵性和读深度信息一致。软件包可在 http://statgene.med.upenn.edu/softprog.html 获得。