Baryshnikova Anastasia, VanderSluis Benjamin, Costanzo Michael, Myers Chad L, Cha Rita S, Andrews Brenda, Boone Charles
Banting and Best Department of Medical Research, The Donnelly Center for Cellular and Biomolecular Research, University of Toronto, Toronto, Ontario M5S 3E1, Canada.
G3 (Bethesda). 2013 Oct 3;3(10):1741-51. doi: 10.1534/g3.113.007377.
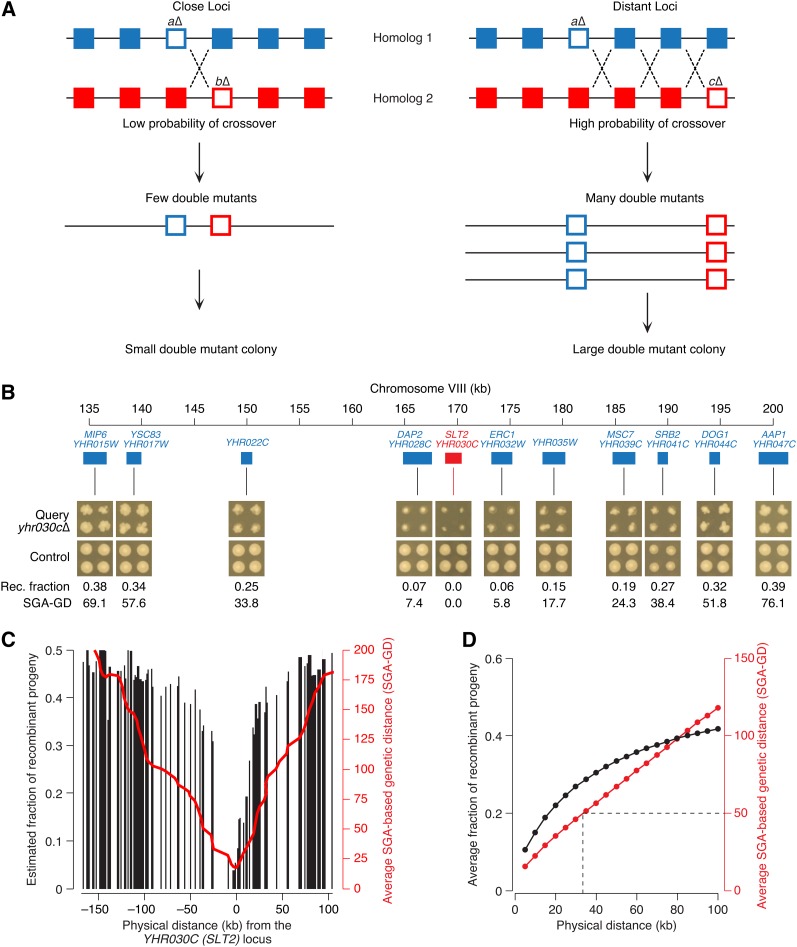
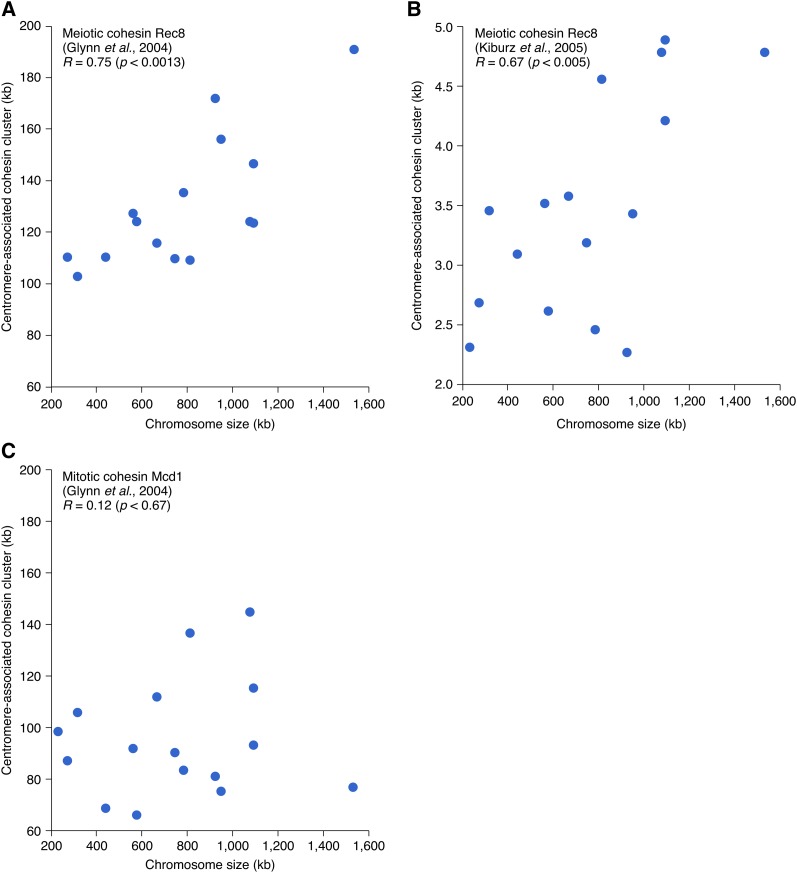
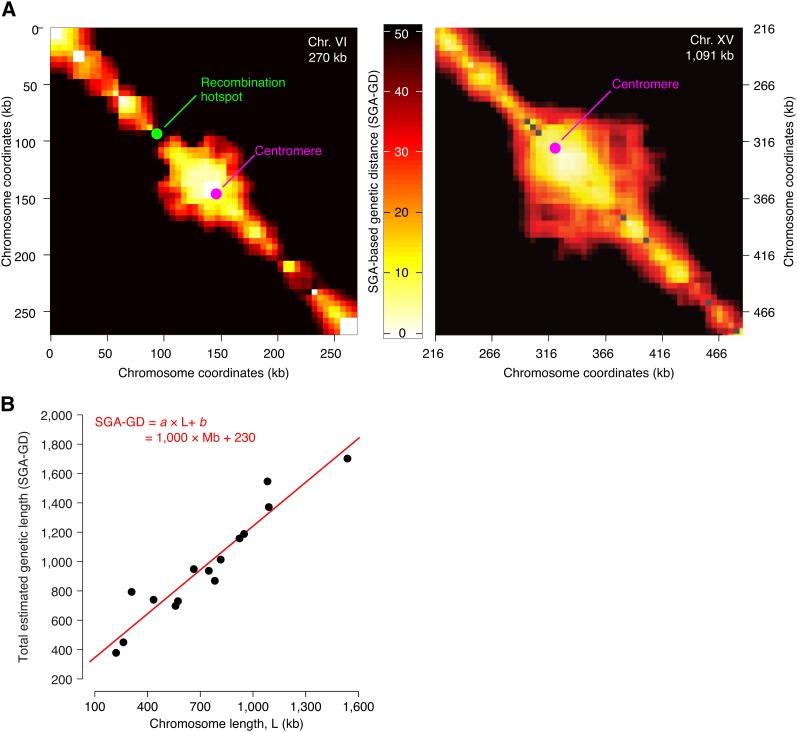
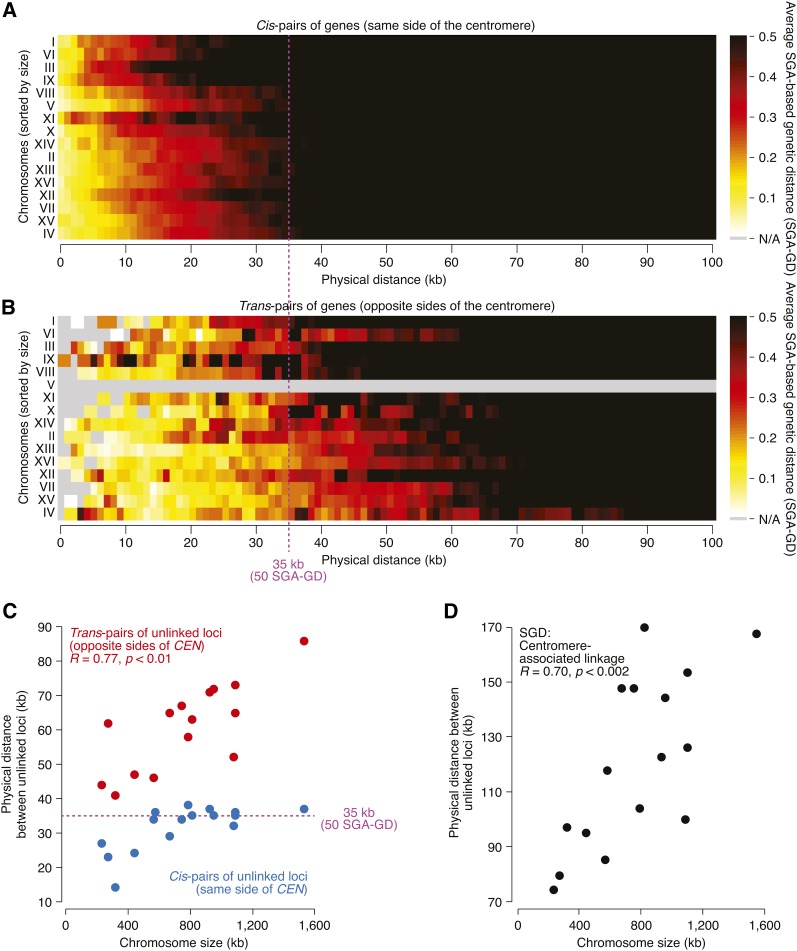
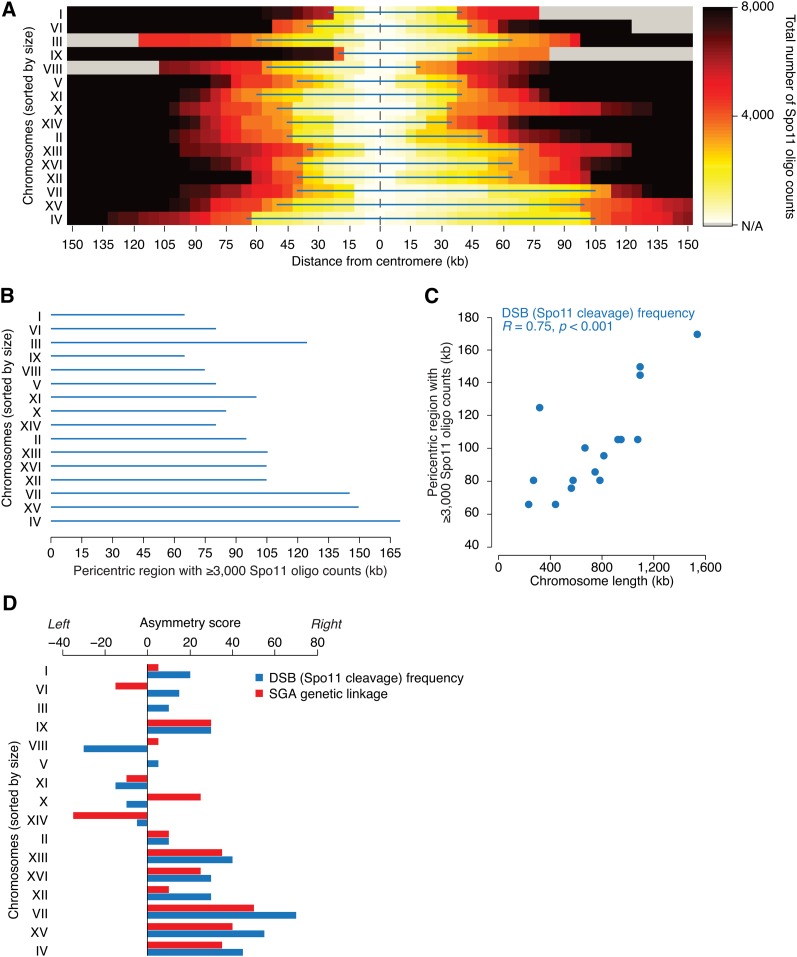
Synthetic genetic array (SGA) analysis automates yeast genetics, enabling high-throughput construction of ordered arrays of double mutants. Quantitative colony sizes derived from SGA analysis can be used to measure cellular fitness and score for genetic interactions, such as synthetic lethality. Here we show that SGA colony sizes also can be used to obtain global maps of meiotic recombination because recombination frequency affects double-mutant formation for gene pairs located on the same chromosome and therefore influences the size of the resultant double-mutant colony. We obtained quantitative colony size data for ~1.2 million double mutants located on the same chromosome and constructed a genome-scale genetic linkage map at ~5 kb resolution. We found that our linkage map is reproducible and consistent with previous global studies of meiotic recombination. In particular, we confirmed that the total number of crossovers per chromosome tends to follow a simple linear model that depends on chromosome size. In addition, we observed a previously unappreciated relationship between the size of linkage regions surrounding each centromere and chromosome size, suggesting that crossovers tend to occur farther away from the centromere on larger chromosomes. The pericentric regions of larger chromosomes also appeared to load larger clusters of meiotic cohesin Rec8, and acquire fewer Spo11-catalyzed DNA double-strand breaks. Given that crossovers too near or too far from centromeres are detrimental to homolog disjunction and increase the incidence of aneuploidy, our data suggest that chromosome size may have a direct role in regulating the fidelity of chromosome segregation during meiosis.
合成遗传阵列(SGA)分析实现了酵母遗传学的自动化,能够高通量构建双突变体的有序阵列。源自SGA分析的定量菌落大小可用于衡量细胞适应性并对遗传相互作用(如合成致死性)进行评分。在此我们表明,SGA菌落大小还可用于获得减数分裂重组的全局图谱,因为重组频率会影响位于同一条染色体上的基因对的双突变体形成,进而影响所得双突变体菌落的大小。我们获得了位于同一条染色体上的约120万个双突变体的定量菌落大小数据,并构建了分辨率约为5 kb的全基因组规模遗传连锁图谱。我们发现我们的连锁图谱具有可重复性,并且与先前关于减数分裂重组的全局研究一致。特别是,我们证实每条染色体的交叉总数倾向于遵循一个依赖于染色体大小的简单线性模型。此外,我们观察到每个着丝粒周围连锁区域的大小与染色体大小之间存在一种先前未被认识到的关系,这表明在较大的染色体上交叉倾向于发生在离着丝粒更远的地方。较大染色体的着丝粒周围区域似乎也装载了更大的减数分裂黏连蛋白Rec8簇,并且获得较少的由Spo11催化的DNA双链断裂。鉴于离着丝粒太近或太远的交叉对同源染色体分离有害并增加非整倍体的发生率,我们的数据表明染色体大小可能在调节减数分裂期间染色体分离保真度方面具有直接作用。