Wasserkort Reinhold, Kalmar Alexandra, Valcz Gabor, Spisak Sandor, Krispin Manuel, Toth Kinga, Tulassay Zsolt, Sledziewski Andrew Z, Molnar Bela
Epigenomics AG, Berlin, Germany.
BMC Cancer. 2013 Aug 30;13:398. doi: 10.1186/1471-2407-13-398.
The septin 9 gene (SEPT9) codes for a GTP-binding protein associated with filamentous structures and cytoskeleton formation. SEPT9 plays a role in multiple cancers as either an oncogene or a tumor suppressor gene. Regulation of SEPT9 expression is complex and not well understood; however, hypermethylation of the gene was recently introduced as a biomarker for early detection of colorectal cancer (CRC) and has been linked to cancer of the breast and of the head and neck. Because the DNA methylation landscape of different regions of SEPT9 is poorly understood in cancer, we analyzed the methylation patterns of this gene in distinct cell populations from normal and diseased colon mucosa.
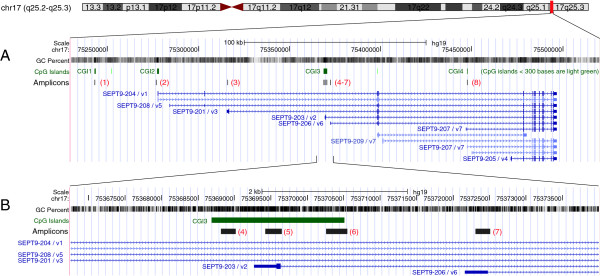
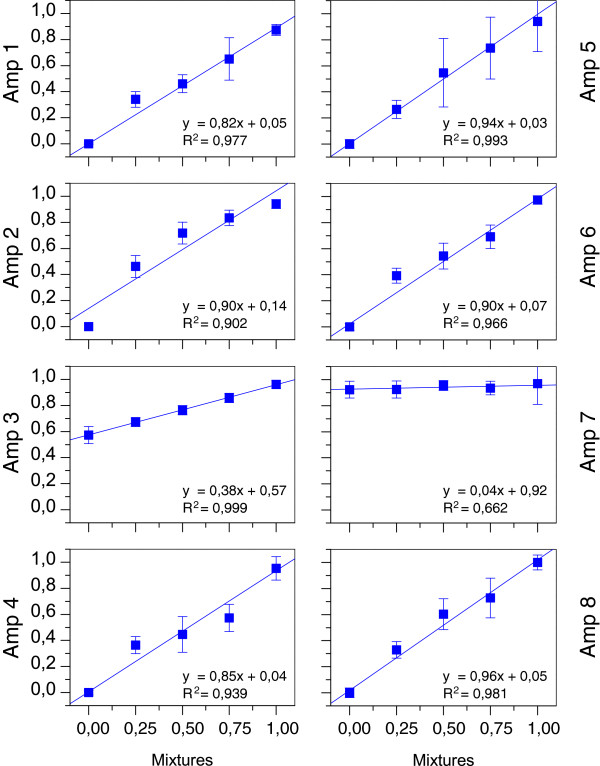
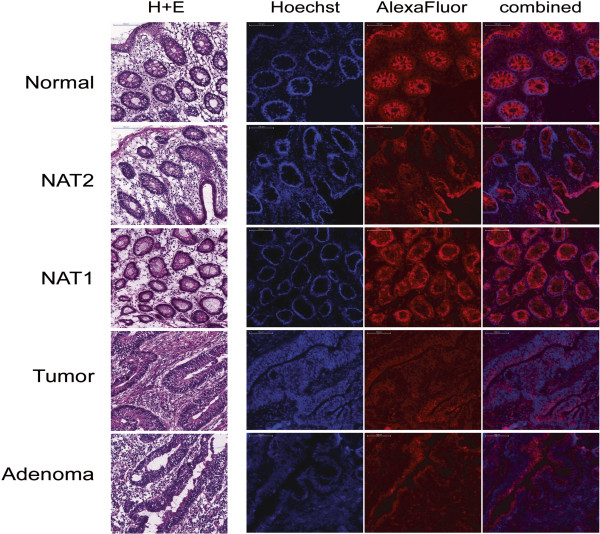
Laser capture microdissection was performed to obtain homogeneous populations of epithelial and stromal cells from normal, adenomatous, and tumorous colon mucosa. Microdissected samples were analyzed using direct bisulfite sequencing to determine the DNA methylation status of eight regions within and near the SEPT9 gene. Septin-9 protein expression was assessed using immunohistochemistry (IHC).
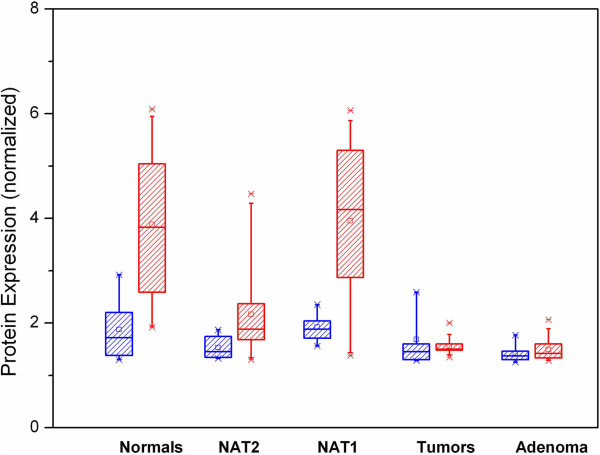
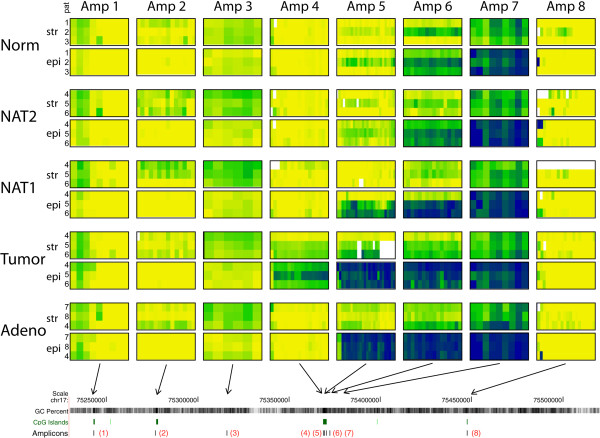
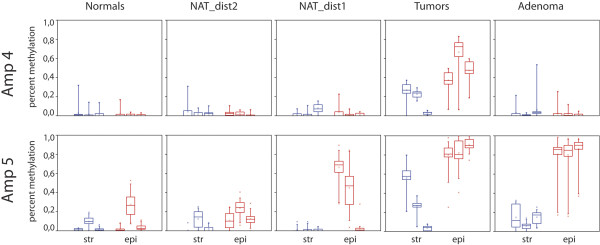
Regions analyzed in SEPT9 were unmethylated in normal tissue except for a methylation boundary detected downstream of the largest CpG island. In adenoma and tumor tissues, epithelial cells displayed markedly increased DNA methylation levels (>80%, p <0.0001) in only one of the CpG islands investigated. SEPT9 methylation in stromal cells increased in adenomatous and tumor tissues (≤50%, p <0.0001); however, methylation did not increase in stromal cells of normal tissue close to the tumor. IHC data indicated a significant decrease (p <0.01) in Septin-9 protein levels in epithelial cells derived from adenoma and tumor tissues; Septin-9 protein levels in stromal cells were low in all tissues.
Hypermethylation of SEPT9 in adenoma and CRC specimens is confined to one of several CpG islands of this gene. Tumor-associated aberrant methylation originates in epithelial cells; stromal cells appear to acquire hypermethylation subsequent to epithelial cells, possibly through field effects. The region in SEPT9 with disease-related hypermethylation also contains the CpGs targeted by a novel blood-based screening test (Epi proColon®), providing further support for the clinical relevance of this biomarker.
septin 9基因(SEPT9)编码一种与丝状结构和细胞骨架形成相关的GTP结合蛋白。SEPT9在多种癌症中作为癌基因或肿瘤抑制基因发挥作用。SEPT9表达的调控较为复杂,目前尚未完全明确;然而,该基因的高甲基化最近被引入作为结直肠癌(CRC)早期检测的生物标志物,并且与乳腺癌以及头颈癌相关。由于在癌症中对SEPT9不同区域的DNA甲基化情况了解甚少,我们分析了该基因在正常和病变结肠黏膜不同细胞群体中的甲基化模式。
采用激光捕获显微切割技术从正常、腺瘤性和肿瘤性结肠黏膜中获取上皮细胞和基质细胞的均一群体。对显微切割后的样本进行直接亚硫酸氢盐测序分析,以确定SEPT9基因内部及附近八个区域的DNA甲基化状态。使用免疫组织化学(IHC)评估septin-9蛋白表达。
在正常组织中,除了在最大的CpG岛下游检测到的甲基化边界外,SEPT9中分析的区域均未甲基化。在腺瘤和肿瘤组织中,上皮细胞仅在其中一个研究的CpG岛中显示出显著增加的DNA甲基化水平(>80%,p<0.0001)。基质细胞中的SEPT9甲基化在腺瘤和肿瘤组织中增加(≤50%,p<0.0001);然而,靠近肿瘤的正常组织基质细胞中的甲基化并未增加。免疫组织化学数据表明,来自腺瘤和肿瘤组织的上皮细胞中septin-9蛋白水平显著降低(p<0.01);所有组织中基质细胞的septin-9蛋白水平均较低。
腺瘤和CRC标本中SEPT9的高甲基化局限于该基因的几个CpG岛之一。肿瘤相关的异常甲基化起源于上皮细胞;基质细胞似乎在上皮细胞之后获得高甲基化,可能是通过场效应。SEPT9中与疾病相关的高甲基化区域也包含一种新型血液筛查检测(Epi proColon®)所针对的CpG,这为该生物标志物的临床相关性提供了进一步支持。