Seo Jeong Kee
Division of Pediatric Gastroenterology, Hepatology and Nutrition, Department of Pediatrics, College of Medicine, Seoul National University, Seoul, Korea.
Pediatr Gastroenterol Hepatol Nutr. 2012 Dec;15(4):197-209. doi: 10.5223/pghn.2012.15.4.197. Epub 2012 Dec 31.
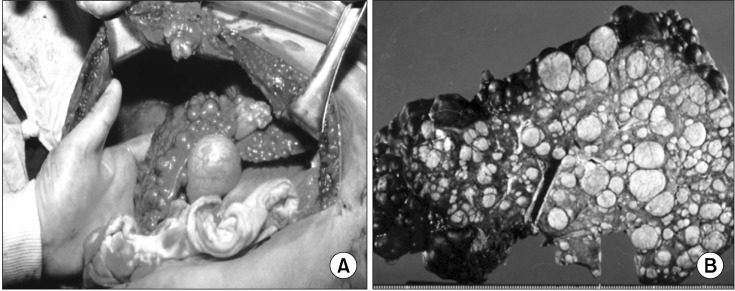
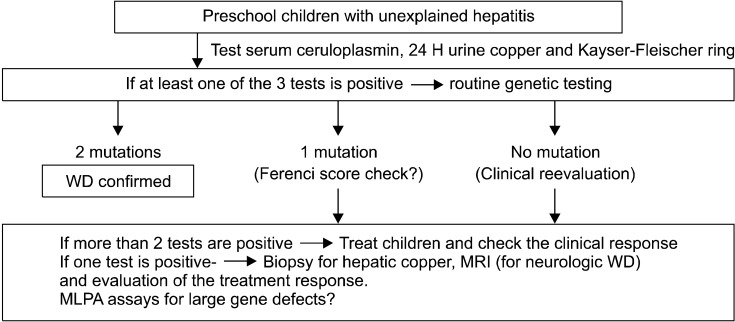
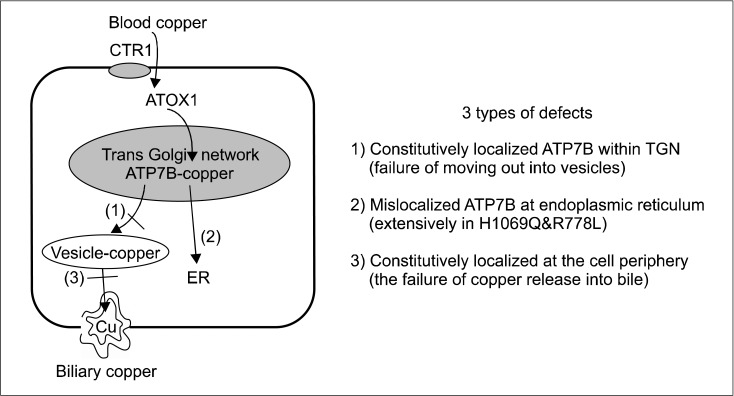
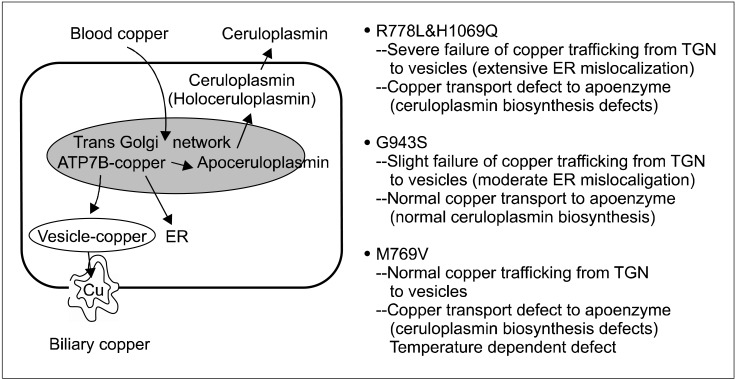
Wilson disease (WD) is an autosomal recessive disorder of copper metabolism that results in accumulation of copper primarily in the liver, brain and cornea. Mutations in the WD gene, ATP7B, cause failure of copper excretion from hepatocyte into bile and a defective synthesis of ceruloplasmin. More than 500 mutations are now recognized, scattered throughout the ATP7B gene. Since WD has protean clinical presentations, awareness of WD in clinical practice is important for the early diagnosis and prevention of accumulated copper toxicity. Molecular genetic testing is playing an increasingly important role in the diagnosis of WD in uncertain cases and family screening. Siblings should be screened for WD once an index case has been diagnosed. Discrimination of heterozygotes from asymptomatic patients is essential to avoid inappropriate lifelong therapy for heterozygotes. Genetic testing, either by haplotype analysis or by mutation analysis, is the only definite solution for differentiating heterozygote carriers from affected asymptomatic patients. Routine genetic testing, because of the multitude of documented mutations, has been thought to be impractical until recently. However, genetic testing is now being more actively applied to the diagnosis of WD, particularly in young children in whom conventional biochemical diagnosis has much limitation and only genetic testing is able to confirm WD. Because advancement of modern biochemical technology now allows more rapid, easier, and less expensive mutation detection, direct DNA sequencing could be actively considered as the primary mode of diagnostic investigation rather than a supplementary test to the conventional biochemical tests. This review will focus on the recent advancement of molecular genetics and genetic diagnosis of WD in very young children on the basis of research data of the Seoul National University Children's Hospital and recent literature.
威尔逊病(WD)是一种常染色体隐性铜代谢紊乱疾病,主要导致铜在肝脏、大脑和角膜中蓄积。WD基因ATP7B发生突变,会致使肝细胞向胆汁中排泄铜失败以及铜蓝蛋白合成缺陷。目前已识别出500多种突变,遍布于整个ATP7B基因。由于WD临床表现多样,临床实践中对WD的认识对于早期诊断和预防铜蓄积毒性至关重要。分子遗传学检测在不明病例的WD诊断及家族筛查中发挥着越来越重要的作用。一旦确诊索引病例,就应对其兄弟姐妹进行WD筛查。区分杂合子与无症状患者对于避免对杂合子进行不恰当的终身治疗至关重要。通过单倍型分析或突变分析进行基因检测,是区分杂合子携带者与无症状患病患者的唯一确切方法。由于已记录的突变众多,直到最近,常规基因检测一直被认为不切实际。然而,基因检测现在正更积极地应用于WD的诊断,尤其是在常规生化诊断有很大局限性且只有基因检测能够确诊WD的幼儿中。由于现代生化技术的进步现在允许更快速、更容易且更便宜地进行突变检测,直接DNA测序可被积极视为诊断研究的主要模式,而不是常规生化检测的补充检测。本综述将基于首尔国立大学儿童医院的研究数据和近期文献,重点关注幼儿WD分子遗传学和基因诊断的最新进展。