Measurement Science and Standards, National Research Council Canada, Ottawa, Ontario, Canada.
PLoS One. 2013 Aug 30;8(8):e73499. doi: 10.1371/journal.pone.0073499. eCollection 2013.
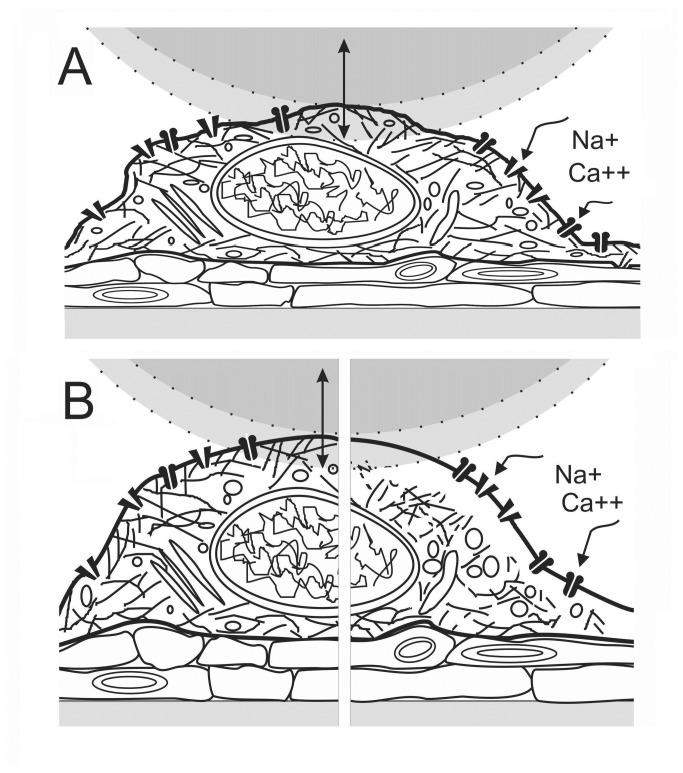
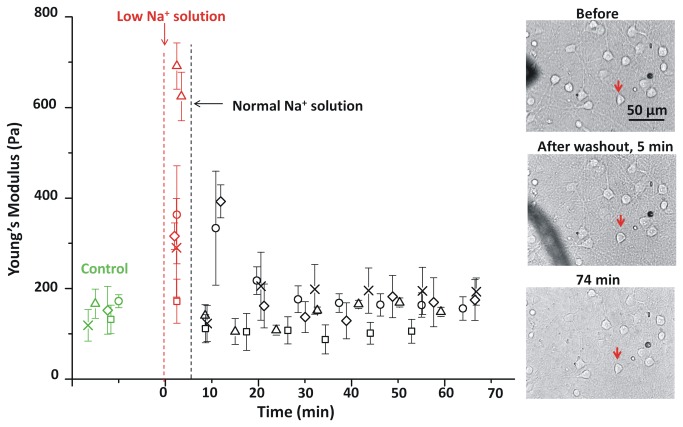
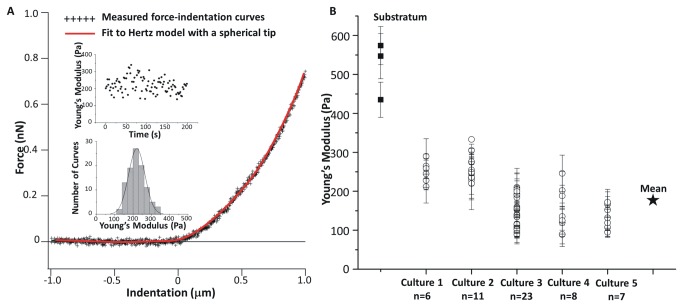
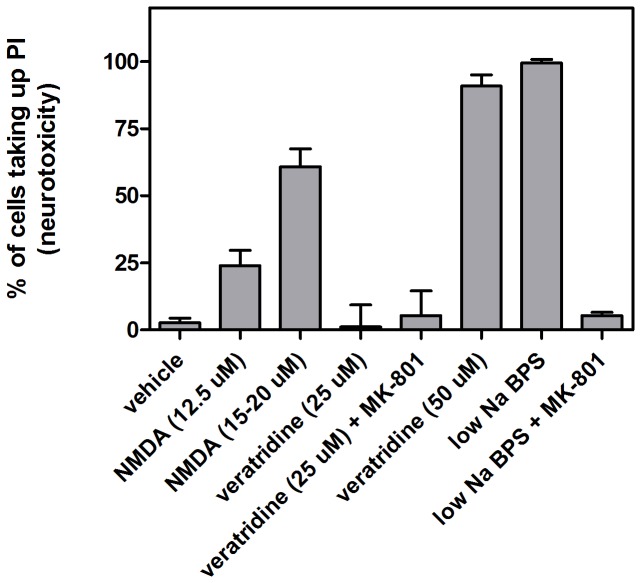
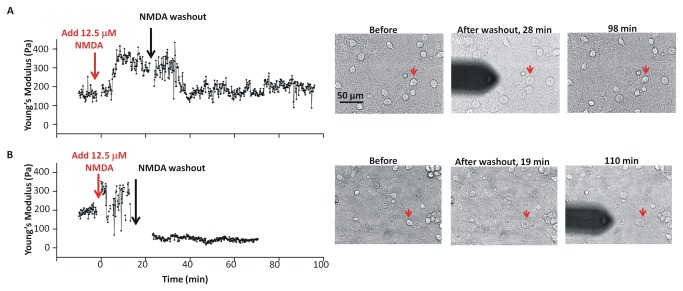
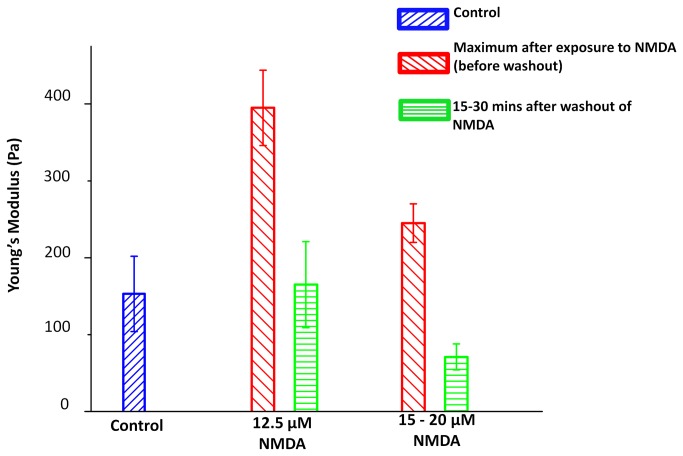
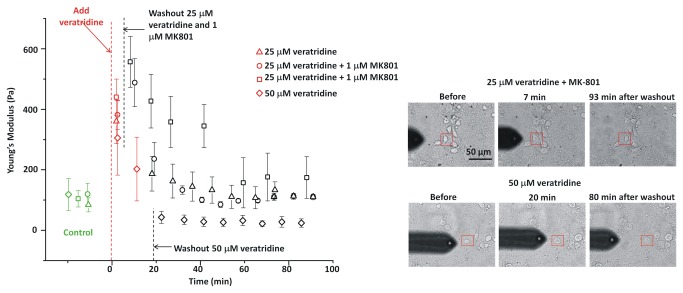
In ischemic and traumatic brain injury, hyperactivated glutamate (N-methyl-D-aspartic acid, NMDA) and sodium (Nav) channels trigger excitotoxic neuron death. Na(+), Ca(++) and H2O influx into affected neurons elicits swelling (increased cell volume) and pathological blebbing (disassociation of the plasma membrane's bilayer from its spectrin-actomyosin matrix). Though usually conflated in injured tissue, cell swelling and blebbing are distinct processes. Around an injury core, salvageable neurons could be mildly swollen without yet having suffered the bleb-type membrane damage that, by rendering channels leaky and pumps dysfunctional, exacerbates the excitotoxic positive feedback spiral. Recognizing when neuronal inflation signifies non-lethal osmotic swelling versus blebbing should further efforts to salvage injury-penumbra neurons. To assess whether the mechanical properties of osmotically-swollen versus excitotoxically-blebbing neurons might be cytomechanically distinguishable, we measured cortical neuron elasticity (gauged via atomic force microscopy (AFM)-based force spectroscopy) upon brief exposure to hypotonicity or to excitotoxic agonists (glutamate and Nav channel activators, NMDA and veratridine). Though unperturbed by solution exchange per se, elasticity increased abruptly with hypotonicity, with NMDA and with veratridine. Neurons then invariably softened towards or below the pre-treatment level, sometimes starting before the washout. The initial channel-mediated stiffening bespeaks an abrupt elevation of hydrostatic pressure linked to NMDA or Nav channel-mediated ion/H2O fluxes, together with increased [Ca(++)]int-mediated submembrane actomyosin contractility. The subsequent softening to below-control levels is consistent with the onset of a lethal level of bleb damage. These findings indicate that dissection/identification of molecular events during the excitotoxic transition from stiff/swollen to soft/blebbing is warranted and should be feasible.
在缺血性和外伤性脑损伤中,过度激活的谷氨酸(N-甲基-D-天冬氨酸,NMDA)和钠(Nav)通道引发兴奋性毒性神经元死亡。Na(+)、Ca(++) 和 H2O 流入受影响的神经元会引起肿胀(细胞体积增加)和病理性起泡(质膜双层与其血影蛋白-肌动球蛋白基质分离)。尽管在受损组织中通常会混淆,但细胞肿胀和起泡是不同的过程。在损伤核心周围,可挽救的神经元可能会轻微肿胀,而尚未遭受起泡型膜损伤,这种损伤会使通道渗漏和泵功能失调,从而加剧兴奋性正反馈螺旋。认识到神经元膨胀是否表示非致死性渗透压肿胀与起泡,可以进一步努力挽救损伤半影区神经元。为了评估渗透压肿胀的神经元与兴奋性起泡的神经元的力学特性是否在细胞力学上可区分,我们通过原子力显微镜(AFM)基于力谱学测量了皮质神经元的弹性(通过原子力显微镜(AFM)测量)短暂暴露于低渗性或兴奋性激动剂(谷氨酸和 Nav 通道激动剂,NMDA 和藜芦碱)。尽管溶液交换本身不会受到干扰,但渗透压会突然增加,NMDA 和藜芦碱也会增加。然后,神经元总是变软,朝向或低于预处理水平,有时在冲洗之前就开始了。最初的通道介导的变硬表明与 NMDA 或 Nav 通道介导的离子/H2O 通量相关的静水压力突然升高,以及增加的[Ca(++)]int 介导的亚膜肌动球蛋白收缩性。随后软化至低于对照水平与致命水平的起泡损伤的发生一致。这些发现表明,有必要对兴奋性毒性从僵硬/肿胀到变软/起泡的过渡过程中的分子事件进行剖析/鉴定,并且应该是可行的。