Department of Biochemistry, University of Calcutta, Kolkata, West Bengal, India.
PLoS One. 2013 Sep 11;8(9):e74067. doi: 10.1371/journal.pone.0074067. eCollection 2013.
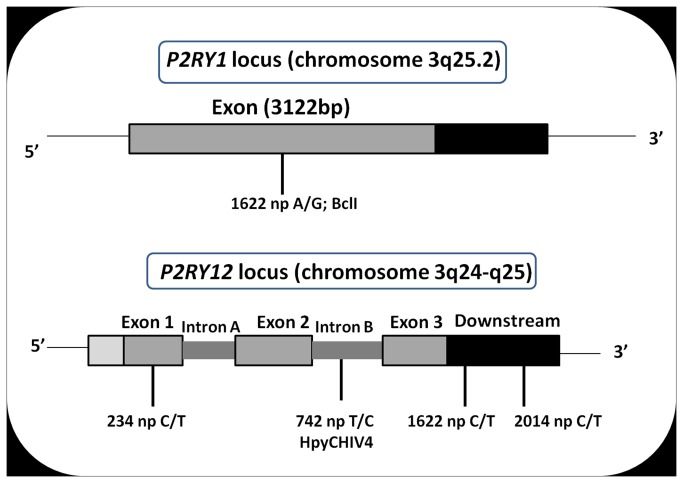
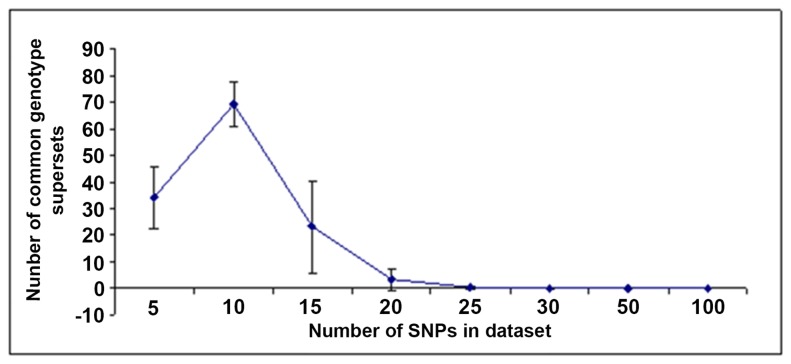
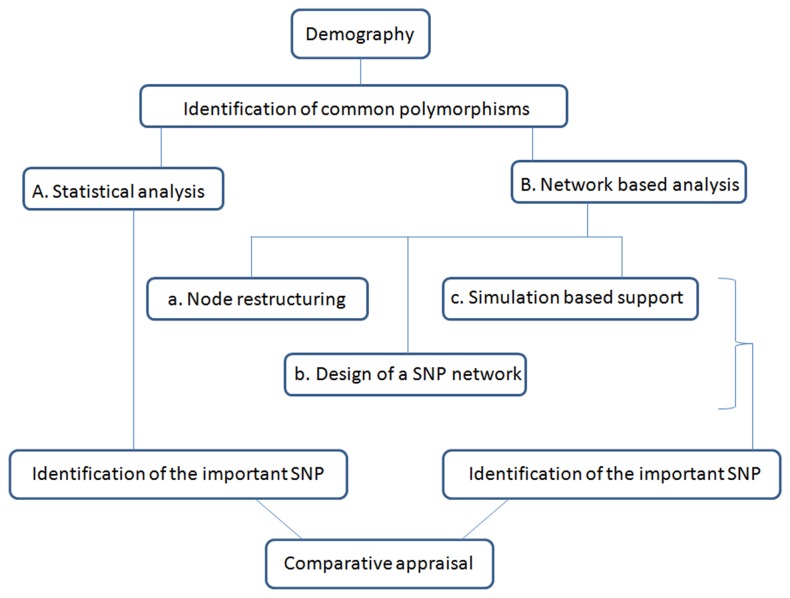
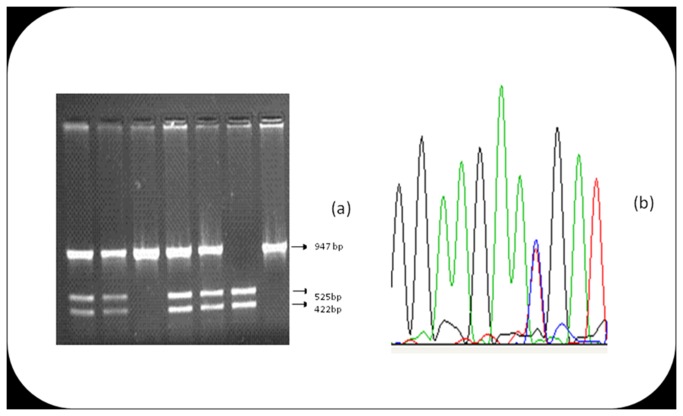
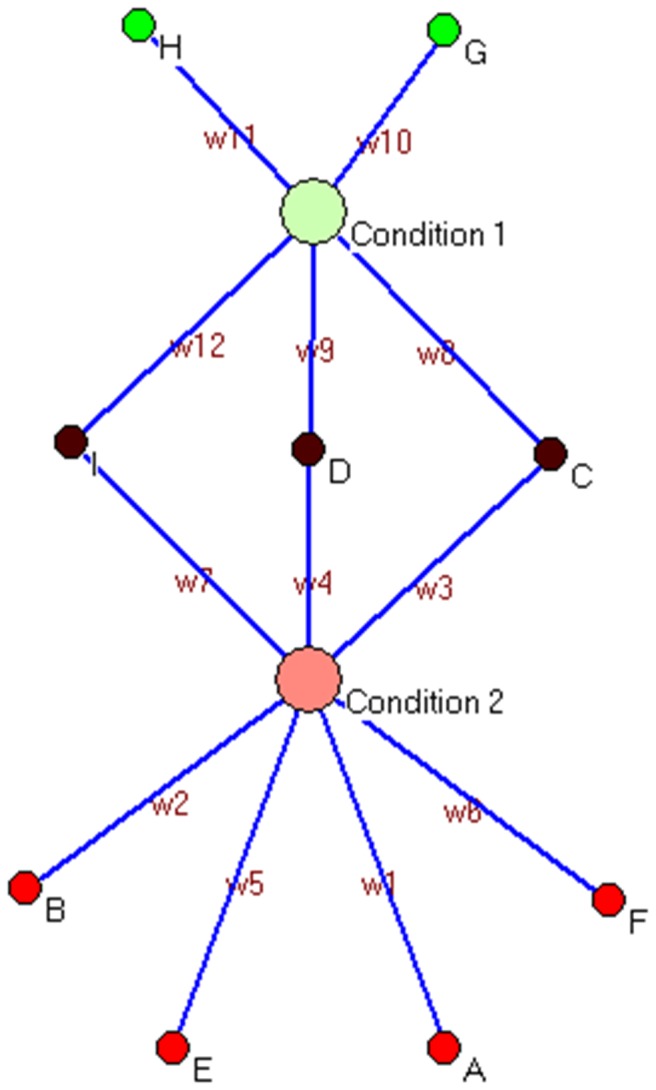
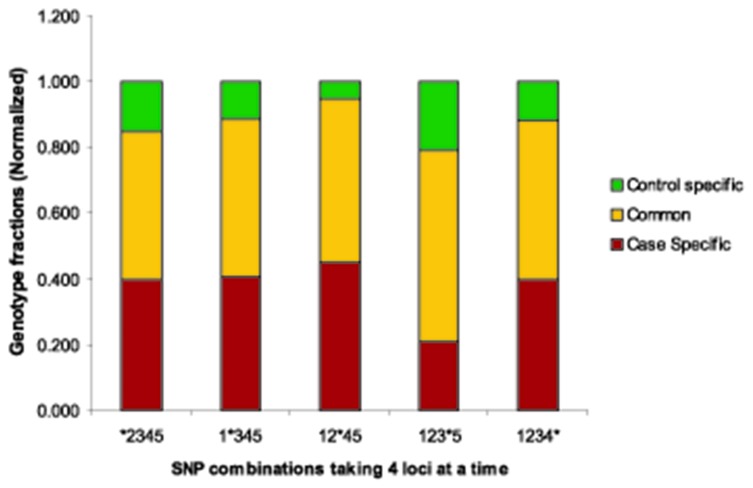
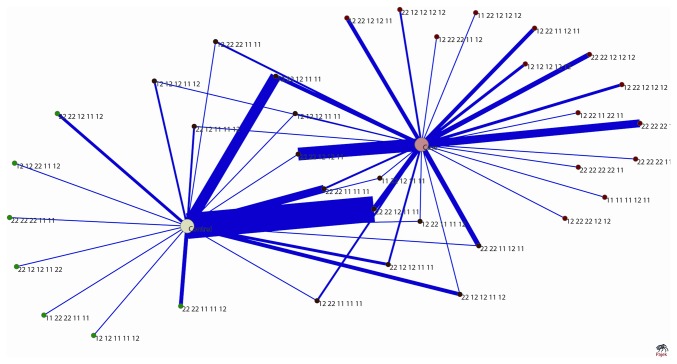
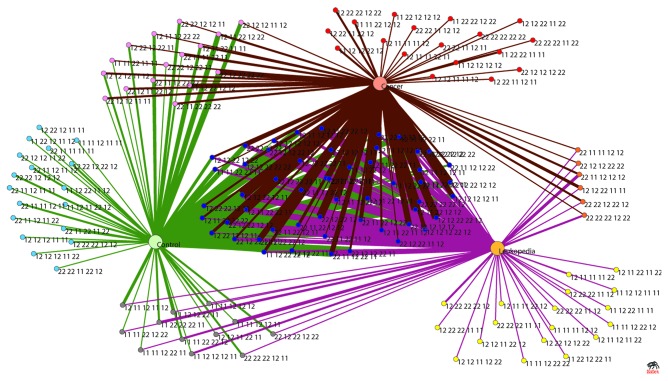
Risk prediction for a particular disease in a population through SNP genotyping exploits tests whose primary goal is to rank the SNPs on the basis of their disease association. This manuscript reveals a different approach of predicting the risk through network representation by using combined genotypic data (instead of a single allele/haplotype). The aim of this study is to classify diseased group and prediction of disease risk by identifying the responsible genotype. Genotypic combination is chosen from five independent loci present on platelet receptor genes P2RY1 and P2RY12. Genotype-sets constructed from combinations of genotypes served as a network input, the network architecture constituting super-nodes (e.g., case and control) and nodes representing individuals, each individual is described by a set of genotypes containing M markers (M = number of SNP). The analysis becomes further enriched when we consider a set of networks derived from the parent network. By maintaining the super-nodes identical, each network is carrying an independent combination of M-1 markers taken from M markers. For each of the network, the ratio of case specific and control specific connections vary and the ratio of super-node specific connection shows variability. This method of network has also been applied in another case-control study which includes oral cancer, precancer and control individuals to check whether it improves presentation and interpretation of data. The analyses reveal a perfect segregation between super-nodes, only a fraction of mixed state being connected to both the super-nodes (i.e. common genotype set). This kind of approach is favorable for a population to classify whether an individual with a particular genotypic combination can be in a risk group to develop disease. In addition with that we can identify the most important polymorphism whose presence or absence in a population can make a large difference in the number of case and control individuals.
通过 SNP 基因分型对人群中特定疾病的风险进行预测,利用的是旨在根据疾病相关性对 SNP 进行排序的测试。本文提出了一种通过网络表示来预测风险的不同方法,该方法使用组合基因型数据(而不是单一等位基因/单倍型)。本研究的目的是通过识别负责的基因型来对疾病组进行分类和预测疾病风险。从血小板受体基因 P2RY1 和 P2RY12 上的五个独立位点中选择基因型组合。由基因型组合构建的基因型集作为网络输入,网络结构由超级节点(例如病例和对照)和代表个体的节点组成,每个个体由一组包含 M 个标记(M=SNP 数量)的基因型描述。当我们考虑从父网络衍生出的一组网络时,分析变得更加丰富。通过保持超级节点相同,每个网络都携带从 M 个标记中选择的 M-1 个独立标记的组合。对于每个网络,病例特异性和对照特异性连接的比例各不相同,超级节点特异性连接的比例显示出可变性。这种网络方法也应用于另一项病例对照研究,其中包括口腔癌、癌前病变和对照个体,以检查它是否改善了数据的呈现和解释。分析揭示了超级节点之间的完美分离,只有一小部分混合状态连接到两个超级节点(即共同基因型集)。这种方法有利于对人群进行分类,以确定具有特定基因型组合的个体是否处于发病风险组。此外,我们还可以确定最重要的多态性,其在人群中的存在或缺失可以在病例和对照个体的数量上产生很大的差异。