Alfadhel Majid, AlShehhi Wafaa, Alshaalan Hesham, Al Balwi Mohammed, Eyaid Wafaa
Dr. Majid Alfadhel . Pediatrics, King Abdulaziz Medical City, Sheikh Jabir Al Ahmed al Sabah Street, Al Rimayah, PO Box 22490, 1510 Riyadh 11426, Saudi Arabia .
Ann Saudi Med. 2013 Jul-Aug;33(4):382-6. doi: 10.5144/0256-4947.2013.382.
Mucolipidosis II (MLII) is characterized by severe global developmental delay, coarse facial features, skeletal deformities, and other systemic involvement. It is caused by a deficiency in N-acetylglucosamine-1 phosphotransferase.
This is a case series study conducted at King Abdulaziz Medical City in Riyadh, Saudi Arabia, between 2008-2012.
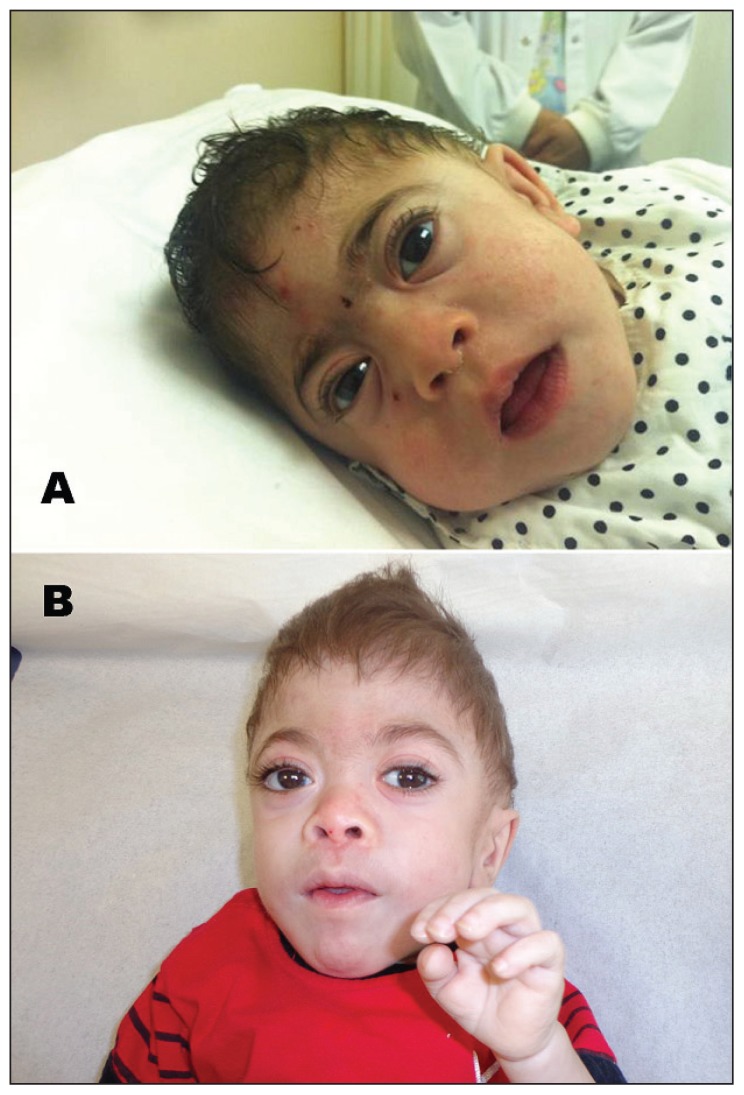
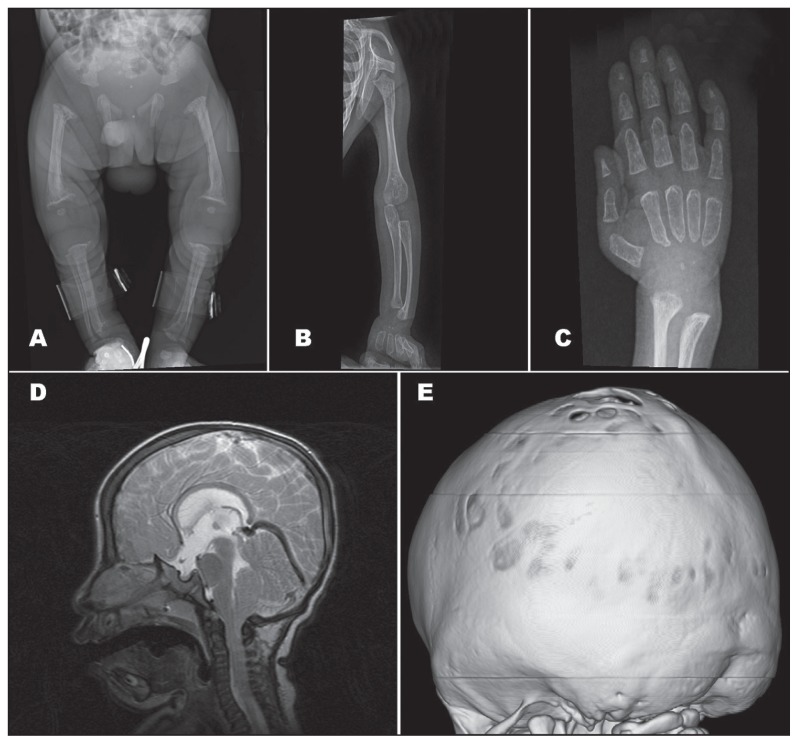
We described three unrelated Saudi children who presented with neonatal hyperparathyroidism, microcephaly, craniosynostosis, coarse facial features, cardiac involvement, and skeletal deformities.
The MLII diagnosis was confirmed by assaying enzyme activities in fibroblasts, which showed a severe reduction in hydrolyzed substrates compared to controls, and by identifying a pathogenic homozygous GNPTAB gene mutation. One of the children died at 2 months of age due to severe pulmonary hypertension, and the other two children were still alive at 12 months and 18 months of age, respectively. Both surviving children had severe global developmental delay at 2 months of age.
Clinicians should investigate any child presenting with neonatal hyperparathyroidism, craniosynostosis, skeletal deformities, and coarse facial features for MLII.
II型粘脂贮积症(MLII)的特征为严重的全面发育迟缓、面部特征粗糙、骨骼畸形及其他全身受累情况。它由N-乙酰葡糖胺-1-磷酸转移酶缺乏所致。
这是一项于2008年至2012年在沙特阿拉伯利雅得的阿卜杜勒阿齐兹国王医疗城开展的病例系列研究。
我们描述了三名无血缘关系的沙特儿童,他们表现为新生儿甲状旁腺功能亢进、小头畸形、颅缝早闭、面部特征粗糙、心脏受累及骨骼畸形。
通过检测成纤维细胞中的酶活性(与对照组相比,水解底物严重减少)以及鉴定致病性纯合GNPTAB基因突变,确诊为MLII。其中一名儿童在2个月大时因严重肺动脉高压死亡,另外两名儿童分别在12个月和18个月大时仍存活。两名存活儿童在2个月大时均有严重的全面发育迟缓。
临床医生应对任何出现新生儿甲状旁腺功能亢进、颅缝早闭、骨骼畸形及面部特征粗糙的儿童进行MLII调查。