Integrated Cancer Genomics Division, Translational Genomics Research Institute, Phoenix, AZ, 85004, USA, Neurogenomics Division, Translational Genomics Research Institute, Phoenix, AZ, 85004, USA and Cancer Treatment Centers of America, Medical Oncology, Goodyear, AZ, 85338, USA.
Nucleic Acids Res. 2014 Jan;42(2):e8. doi: 10.1093/nar/gkt865. Epub 2013 Sep 25.
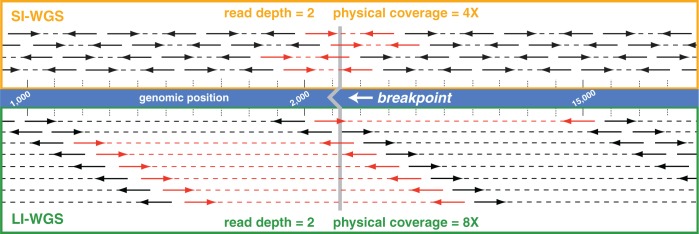
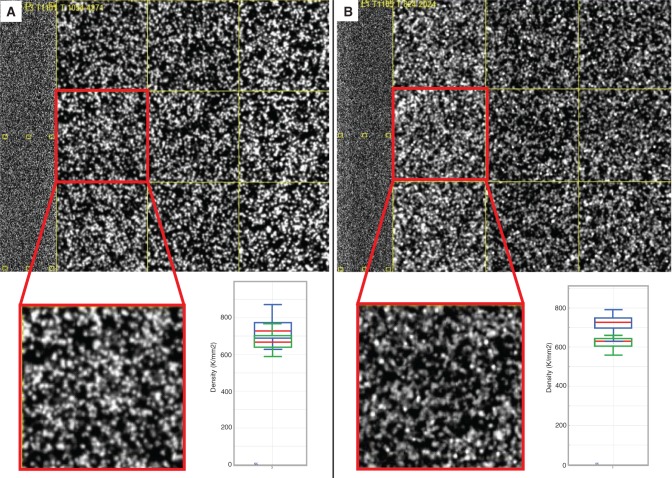
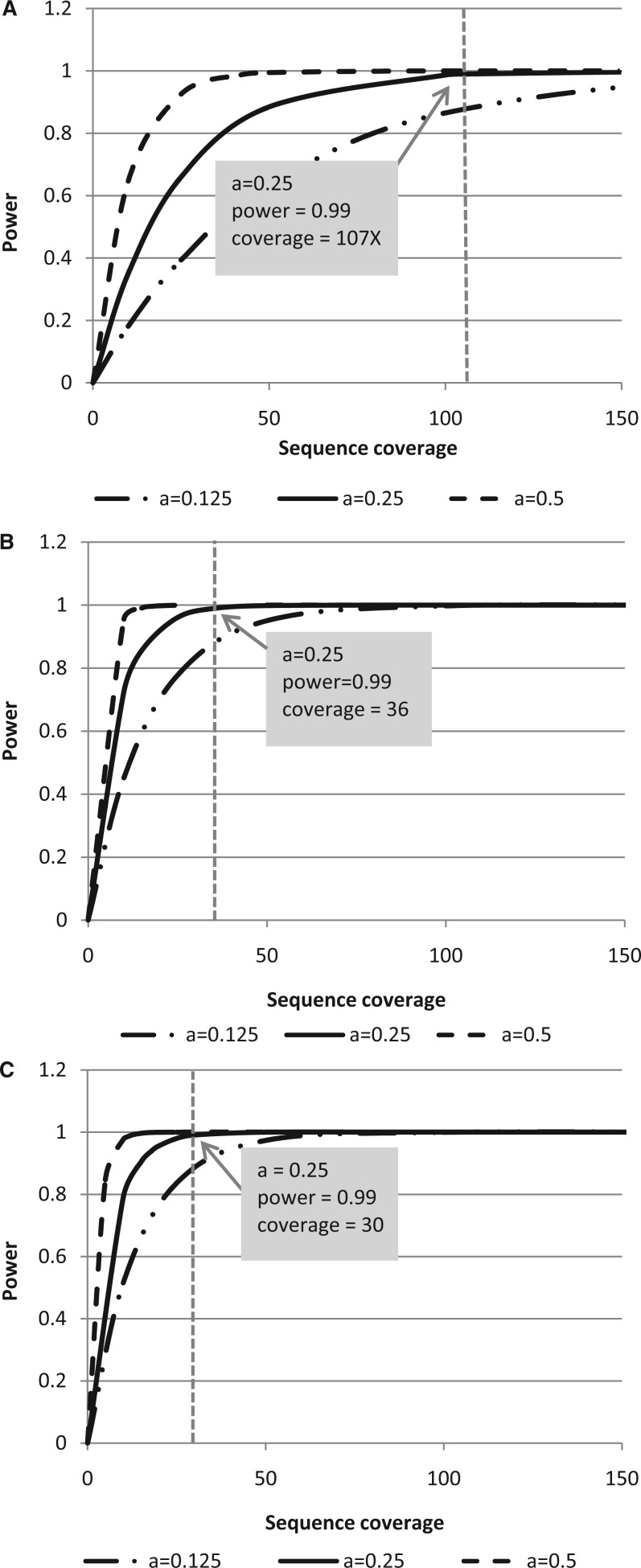
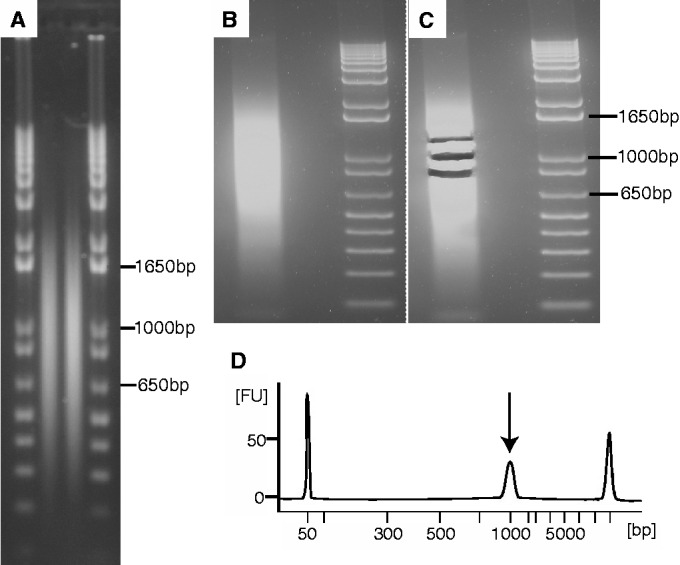
As next-generation sequencing continues to have an expanding presence in the clinic, the identification of the most cost-effective and robust strategy for identifying copy number changes and translocations in tumor genomes is needed. We hypothesized that performing shallow whole genome sequencing (WGS) of 900-1000-bp inserts (long insert WGS, LI-WGS) improves our ability to detect these events, compared with shallow WGS of 300-400-bp inserts. A priori analyses show that LI-WGS requires less sequencing compared with short insert WGS to achieve a target physical coverage, and that LI-WGS requires less sequence coverage to detect a heterozygous event with a power of 0.99. We thus developed an LI-WGS library preparation protocol based off of Illumina's WGS library preparation protocol and illustrate the feasibility of performing LI-WGS. We additionally applied LI-WGS to three separate tumor/normal DNA pairs collected from patients diagnosed with different cancers to demonstrate our application of LI-WGS on actual patient samples for identification of somatic copy number alterations and translocations. With the evolution of sequencing technologies and bioinformatics analyses, we show that modifications to current approaches may improve our ability to interrogate cancer genomes.
随着下一代测序技术在临床上的应用不断扩大,需要确定在肿瘤基因组中识别拷贝数变化和易位的最具成本效益和稳健的策略。我们假设,与浅测序 300-400-bp 插入物(短插入物 WGS)相比,对 900-1000-bp 插入物(长插入物 WGS,LI-WGS)进行浅全基因组测序可以提高我们检测这些事件的能力。先验分析表明,与短插入物 WGS 相比,LI-WGS 实现目标物理覆盖所需的测序量更少,并且 LI-WGS 检测杂合事件所需的序列覆盖更少,检测效能为 0.99。因此,我们基于 Illumina 的 WGS 文库制备方案开发了一种 LI-WGS 文库制备方案,并说明了进行 LI-WGS 的可行性。我们还将 LI-WGS 应用于从不同癌症患者中收集的三个单独的肿瘤/正常 DNA 对,以证明我们在实际患者样本中应用 LI-WGS 来识别体细胞拷贝数改变和易位。随着测序技术和生物信息学分析的发展,我们表明对当前方法的修改可能会提高我们研究癌症基因组的能力。