Boston College, Boston, Chestnut Hill, MA, USA.
BMC Bioinformatics. 2012 Nov 17;13:305. doi: 10.1186/1471-2105-13-305.
DNA capture technologies combined with high-throughput sequencing now enable cost-effective, deep-coverage, targeted sequencing of complete exomes. This is well suited for SNP discovery and genotyping. However there has been little attention devoted to Copy Number Variation (CNV) detection from exome capture datasets despite the potentially high impact of CNVs in exonic regions on protein function.
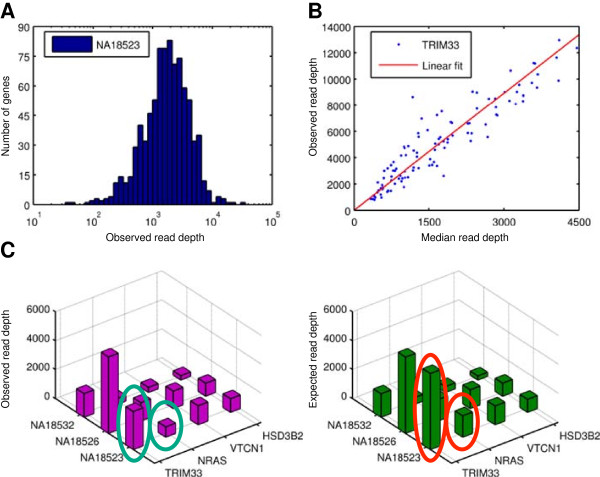
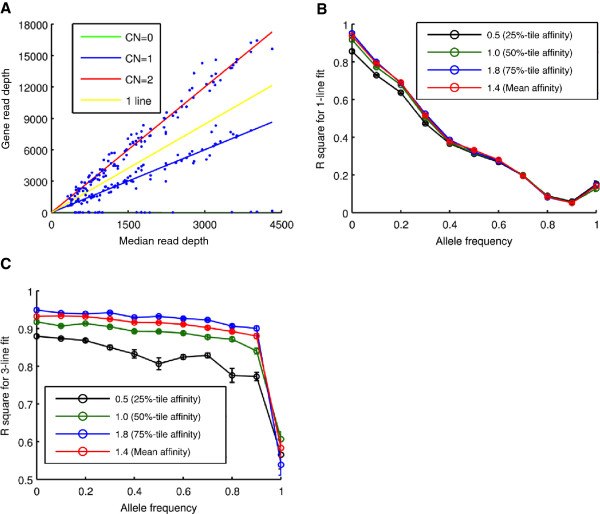
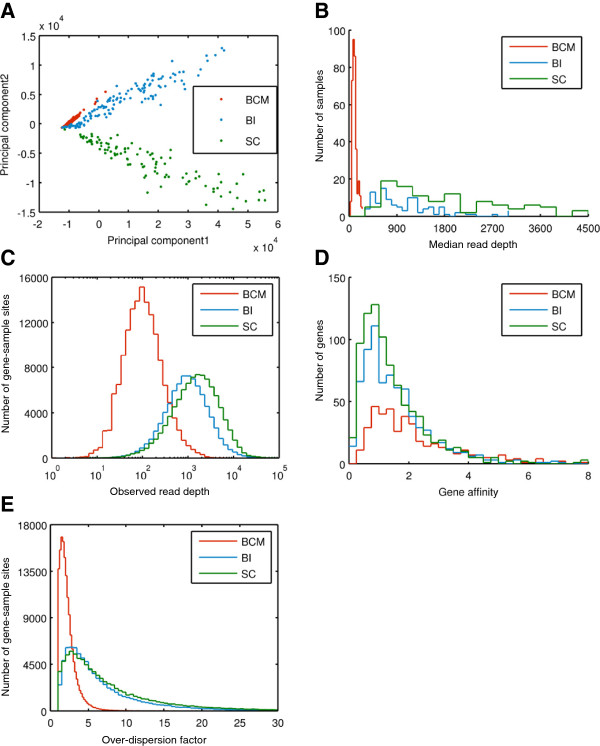
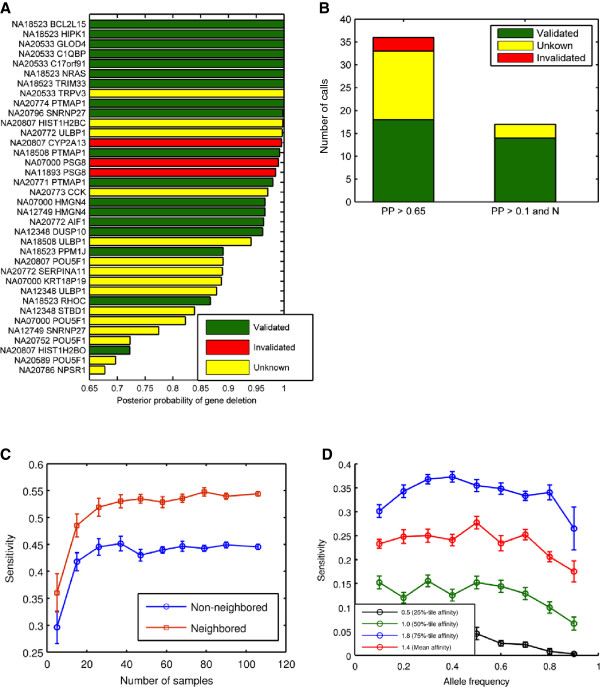
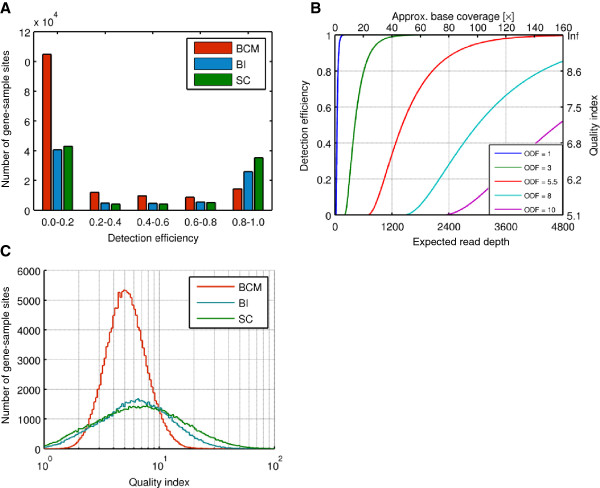
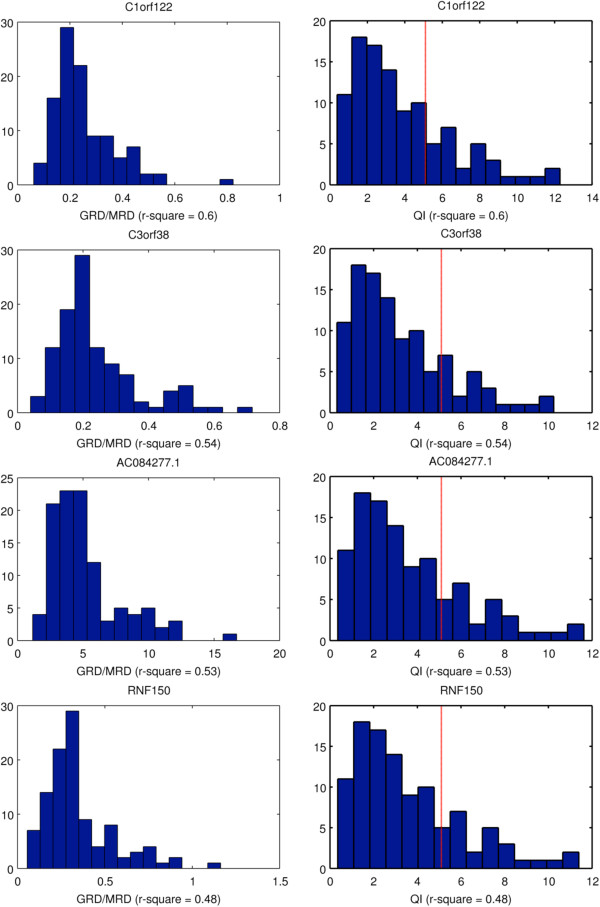
As members of the 1000 Genomes Project analysis effort, we investigated 697 samples in which 931 genes were targeted and sampled with 454 or Illumina paired-end sequencing. We developed a rigorous Bayesian method to detect CNVs in the genes, based on read depth within target regions. Despite substantial variability in read coverage across samples and targeted exons, we were able to identify 107 heterozygous deletions in the dataset. The experimentally determined false discovery rate (FDR) of the cleanest dataset from the Wellcome Trust Sanger Institute is 12.5%. We were able to substantially improve the FDR in a subset of gene deletion candidates that were adjacent to another gene deletion call (17 calls). The estimated sensitivity of our call-set was 45%.
This study demonstrates that exonic sequencing datasets, collected both in population based and medical sequencing projects, will be a useful substrate for detecting genic CNV events, particularly deletions. Based on the number of events we found and the sensitivity of the methods in the present dataset, we estimate on average 16 genic heterozygous deletions per individual genome. Our power analysis informs ongoing and future projects about sequencing depth and uniformity of read coverage required for efficient detection.
DNA 捕获技术与高通量测序的结合使对完整外显子进行经济高效、深度覆盖、靶向测序成为可能。这非常适合 SNP 发现和基因分型。然而,尽管外显子区域的 CNV 对蛋白质功能有潜在的重大影响,但从外显子捕获数据集中检测拷贝数变异(CNV)的关注甚少。
作为 1000 基因组计划分析工作的成员,我们研究了 697 个样本,其中 931 个基因被靶向并与 454 或 Illumina 配对末端测序一起采样。我们开发了一种严格的贝叶斯方法,基于目标区域内的读取深度来检测基因中的 CNV。尽管样本和靶向外显子之间的读取覆盖率存在很大差异,但我们能够在数据集中识别出 107 个杂合性缺失。来自 Wellcome Trust Sanger 研究所的最干净数据集的实验确定的假发现率(FDR)为 12.5%。我们能够在与另一个基因缺失调用相邻的基因缺失候选者的子集(17 个调用)中大大降低 FDR。我们的调用集的估计灵敏度为 45%。
本研究表明,无论是在基于人群的测序项目还是医学测序项目中收集的外显子测序数据集,都将成为检测基因 CNV 事件(尤其是缺失)的有用底物。基于我们发现的事件数量和本数据集方法的灵敏度,我们估计每个个体基因组平均有 16 个基因杂合性缺失。我们的功率分析为正在进行和未来的项目提供了有关测序深度和读取覆盖率均匀性的信息,以实现有效的检测。