Department of Internal Medicine, Yale Cardiovascular Research Center, Section of Cardiovascular Medicine (R.L., Y.J., W.T., X.Z., J.H., J.Y., K.A.M.), Department of Surgery (Cardiac Surgery) (L.Q., G.T.), and Department of Pharmacology (K.A.M.), Yale University, New Haven, CT.
Circulation. 2013 Oct 29;128(18):2047-57. doi: 10.1161/CIRCULATIONAHA.113.002887. Epub 2013 Sep 27.
Smooth muscle cells (SMCs) are remarkably plastic. Their reversible differentiation is required for growth and wound healing but also contributes to pathologies such as atherosclerosis and restenosis. Although key regulators of the SMC phenotype, including myocardin (MYOCD) and KLF4, have been identified, a unifying epigenetic mechanism that confers reversible SMC differentiation has not been reported.
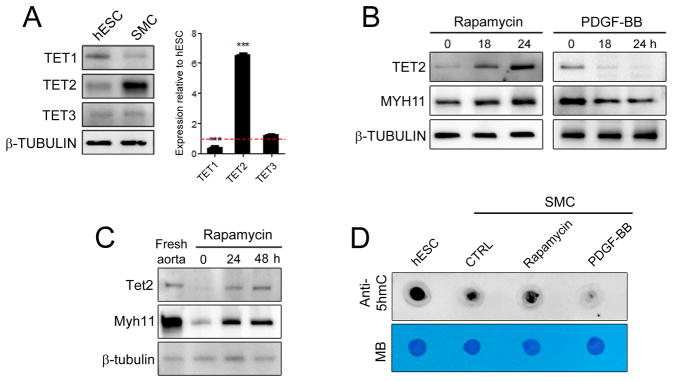
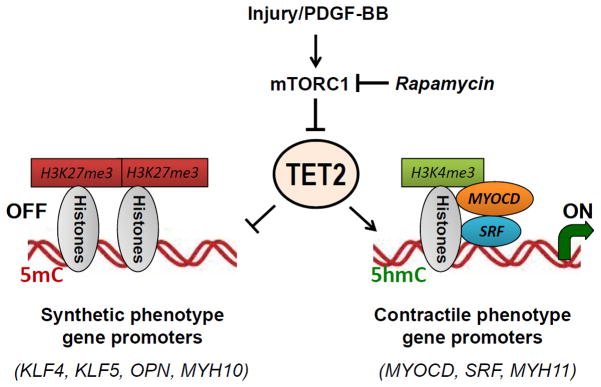
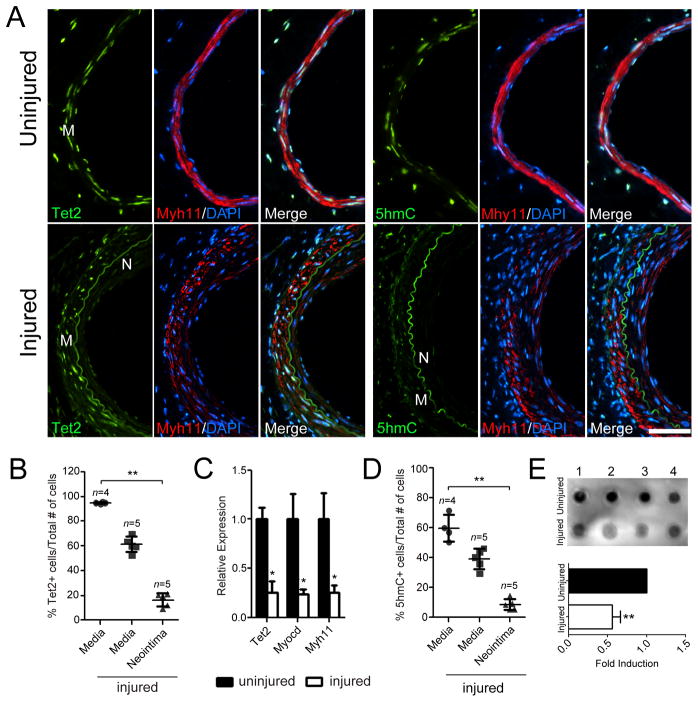
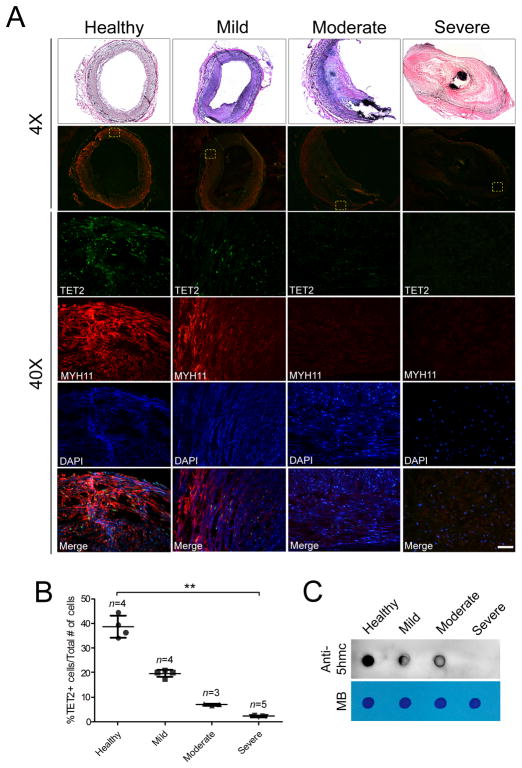
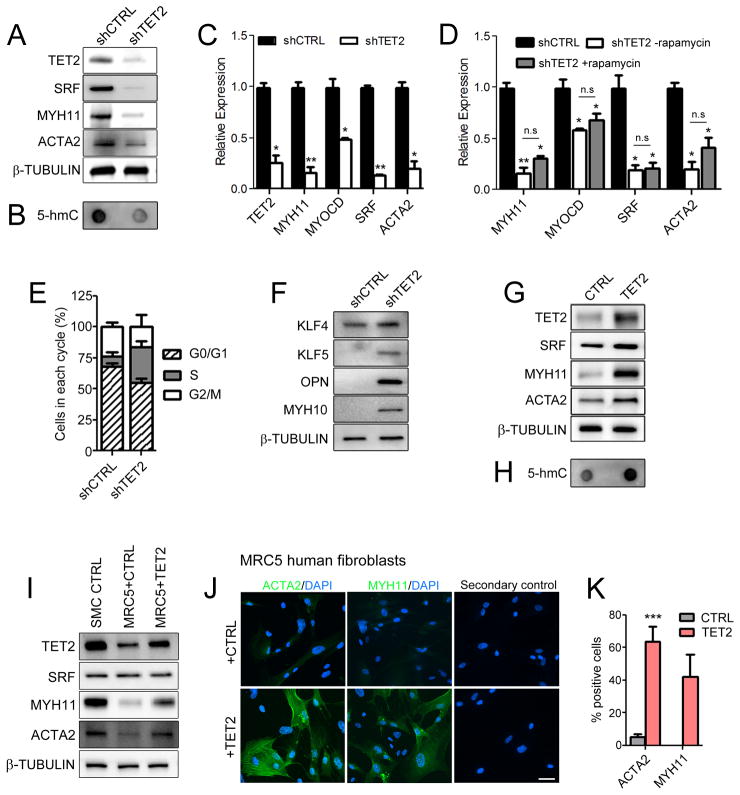
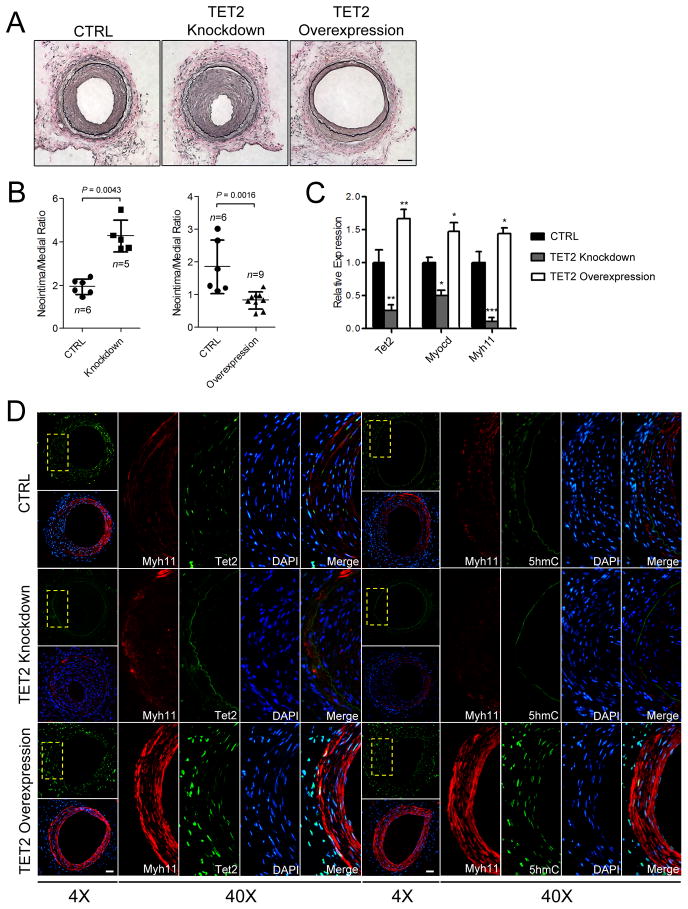
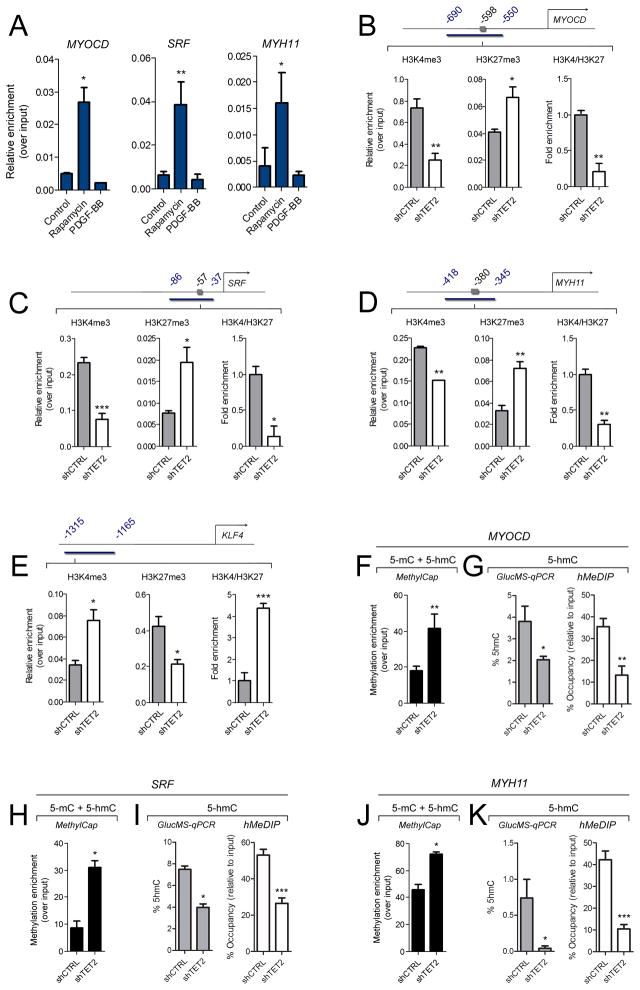
Using human SMCs, human arterial tissue, and mouse models, we report that SMC plasticity is governed by the DNA-modifying enzyme ten-eleven translocation-2 (TET2). TET2 and its product, 5-hydroxymethylcytosine (5-hmC), are enriched in contractile SMCs but reduced in dedifferentiated SMCs. TET2 knockdown inhibits expression of key procontractile genes, including MYOCD and SRF, with concomitant transcriptional upregulation of KLF4. TET2 knockdown prevents rapamycin-induced SMC differentiation, whereas TET2 overexpression is sufficient to induce a contractile phenotype. TET2 overexpression also induces SMC gene expression in fibroblasts. Chromatin immunoprecipitation demonstrates that TET2 coordinately regulates phenotypic modulation through opposing effects on chromatin accessibility at the promoters of procontractile versus dedifferentiation-associated genes. Notably, we find that TET2 binds and 5-hmC is enriched in CArG-rich regions of active SMC contractile promoters (MYOCD, SRF, and MYH11). Loss of TET2 and 5-hmC positively correlates with the degree of injury in murine models of vascular injury and human atherosclerotic disease. Importantly, localized TET2 knockdown exacerbates injury response, and local TET2 overexpression restores the 5-hmC epigenetic landscape and contractile gene expression and greatly attenuates intimal hyperplasia in vivo.
We identify TET2 as a novel and necessary master epigenetic regulator of SMC differentiation.
平滑肌细胞(SMC)具有很强的可塑性。SMC 的可逆分化对于生长和伤口愈合是必需的,但也会导致动脉粥样硬化和再狭窄等病理。虽然已经确定了调节 SMC 表型的关键调节因子,包括肌球蛋白结合蛋白 D(MYOCD)和 KLF4,但尚未报道赋予 SMC 可逆分化的统一表观遗传机制。
使用人 SMC、人动脉组织和小鼠模型,我们报告 SMC 可塑性受 DNA 修饰酶 ten-eleven translocation-2(TET2)的调控。TET2 及其产物 5-羟甲基胞嘧啶(5-hmC)在收缩型 SMC 中丰富,而在去分化的 SMC 中减少。TET2 敲低抑制关键促收缩基因的表达,包括 MYOCD 和 SRF,同时转录上调 KLF4。TET2 敲低可抑制雷帕霉素诱导的 SMC 分化,而 TET2 过表达足以诱导收缩表型。TET2 过表达也可诱导成纤维细胞中的 SMC 基因表达。染色质免疫沉淀表明,TET2 通过对促收缩基因与去分化相关基因启动子的染色质可及性产生相反的影响,协调调节表型调节。值得注意的是,我们发现 TET2 结合并在活跃的 SMC 收缩启动子(MYOCD、SRF 和 MYH11)的富含 CArG 区域富集 5-hmC。TET2 和 5-hmC 的缺失与血管损伤和人类动脉粥样硬化疾病的小鼠模型中的损伤程度呈正相关。重要的是,局部 TET2 敲低会加剧损伤反应,而局部 TET2 过表达恢复 5-hmC 表观遗传景观和收缩基因表达,并大大减轻体内内膜增生。
我们将 TET2 鉴定为 SMC 分化的新型必需主表观遗传调节剂。