Joint BSC-IRB Research Program in Computational Biology, Life Science Department, Barcelona Super computing Center, Barcelona 08034, Spain.
BMC Bioinformatics. 2013 Oct 1;14:286. doi: 10.1186/1471-2105-14-286.
Protein-protein docking, which aims to predict the structure of a protein-protein complex from its unbound components, remains an unresolved challenge in structural bioinformatics. An important step is the ranking of docked poses using a scoring function, for which many methods have been developed. There is a need to explore the differences and commonalities of these methods with each other, as well as with functions developed in the fields of molecular dynamics and homology modelling.
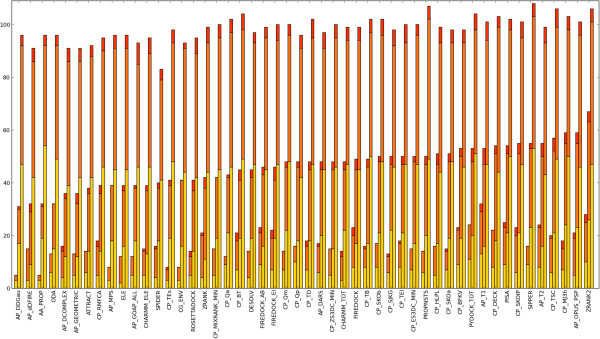
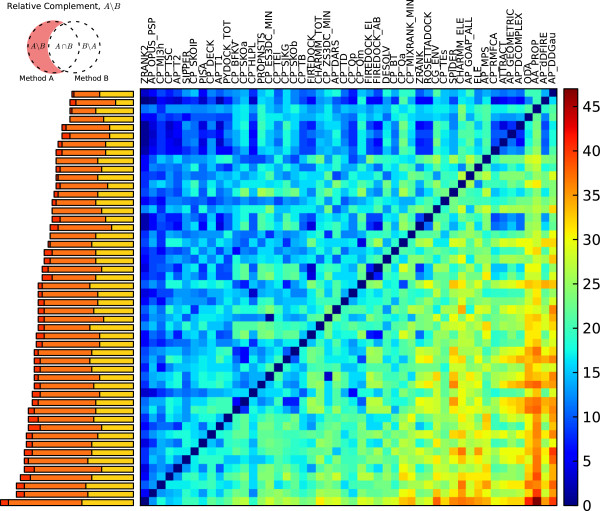
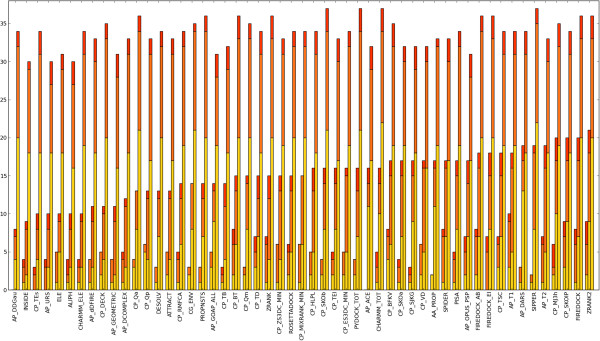
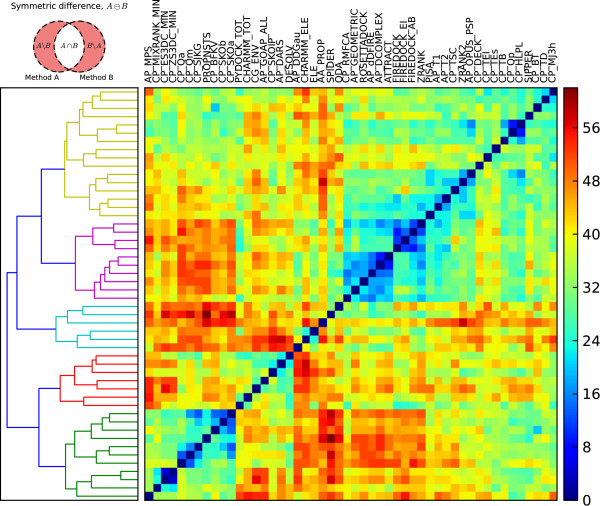
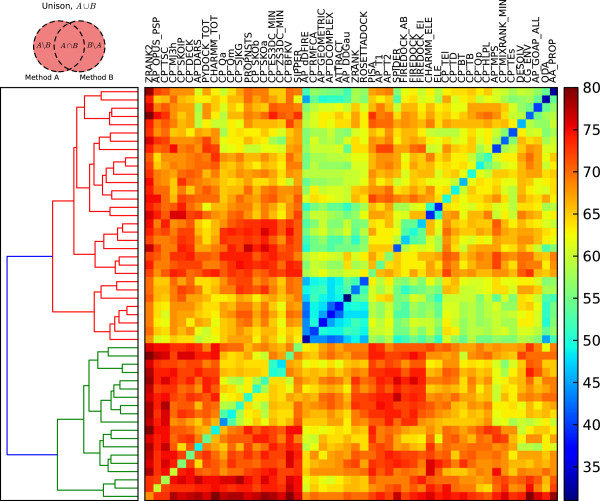
We present an evaluation of 115 scoring functions on an unbound docking decoy benchmark covering 118 complexes for which a near-native solution can be found, yielding top 10 success rates of up to 58%. Hierarchical clustering is performed, so as to group together functions which identify near-natives in similar subsets of complexes. Three set theoretic approaches are used to identify pairs of scoring functions capable of correctly scoring different complexes. This shows that functions in different clusters capture different aspects of binding and are likely to work together synergistically.
All functions designed specifically for docking perform well, indicating that functions are transferable between sampling methods. We also identify promising methods from the field of homology modelling. Further, differential success rates by docking difficulty and solution quality suggest a need for flexibility-dependent scoring. Investigating pairs of scoring functions, the set theoretic measures identify known scoring strategies as well as a number of novel approaches, indicating promising augmentations of traditional scoring methods. Such augmentation and parameter combination strategies are discussed in the context of the learning-to-rank paradigm.
蛋白质-蛋白质对接的目的是根据未结合的成分预测蛋白质-蛋白质复合物的结构,这在结构生物信息学中仍然是一个未解决的挑战。一个重要的步骤是使用评分函数对对接构象进行排序,为此已经开发了许多方法。需要探索这些方法彼此之间以及与分子动力学和同源建模领域开发的功能之间的差异和共同点。
我们评估了 115 种评分函数在未结合对接诱饵基准测试中的表现,该基准测试涵盖了 118 个复合物,其中可以找到近天然解决方案,最高成功率可达 58%。进行了层次聚类,以便将识别类似复合物子集的近天然构象的功能分组在一起。使用三种集合论方法来识别能够正确评分不同复合物的评分函数对。这表明不同聚类中的功能捕获了结合的不同方面,并且可能协同工作。
专门设计用于对接的所有功能表现良好,表明功能在采样方法之间是可转移的。我们还从同源建模领域确定了有前途的方法。此外,根据对接难度和解决方案质量的不同成功率表明需要依赖灵活性的评分。通过对评分函数对进行调查,集合论度量方法确定了已知的评分策略以及许多新的方法,这表明了对传统评分方法的有希望的增强。在学习排序范例的背景下讨论了这种增强和参数组合策略。