Shirali Azam, Stebliankin Vitalii, Karki Ukesh, Shi Jimeng, Chapagain Prem, Narasimhan Giri
Bioinformatics Research Group (BioRG), Knight Foundation School of Computing and Information Sciences, Florida International University, 11200 SW 8th 10 St, Miami, 33199, USA.
Department of Physics, Florida International University, 11200 SW 8th 10 St, Miami, 33199, USA.
BMC Bioinformatics. 2025 Jan 22;26(1):25. doi: 10.1186/s12859-024-05991-4.
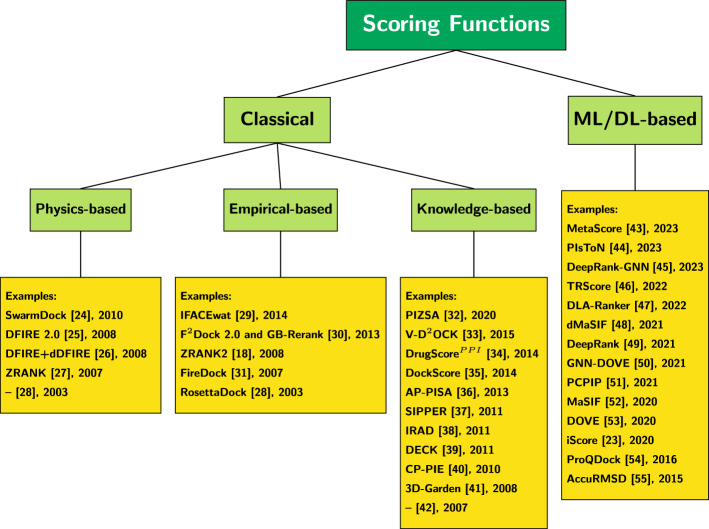
While protein-protein docking is fundamental to our understanding of how proteins interact, scoring protein-protein complex conformations is a critical component of successful docking programs. Without accurate and efficient scoring functions to differentiate between native and non-native binding complexes, the accuracy of current docking tools cannot be guaranteed. Although many innovative scoring functions have been proposed, a good scoring function for docking remains elusive. Deep learning models offer alternatives to using explicit empirical or mathematical functions for scoring protein-protein complexes.
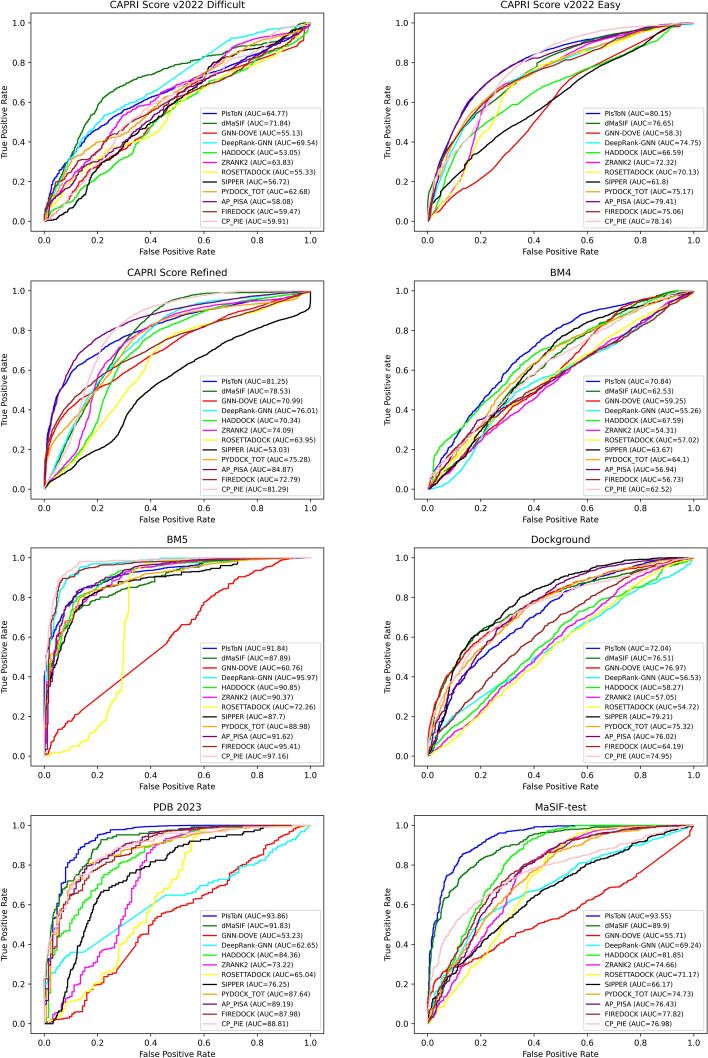
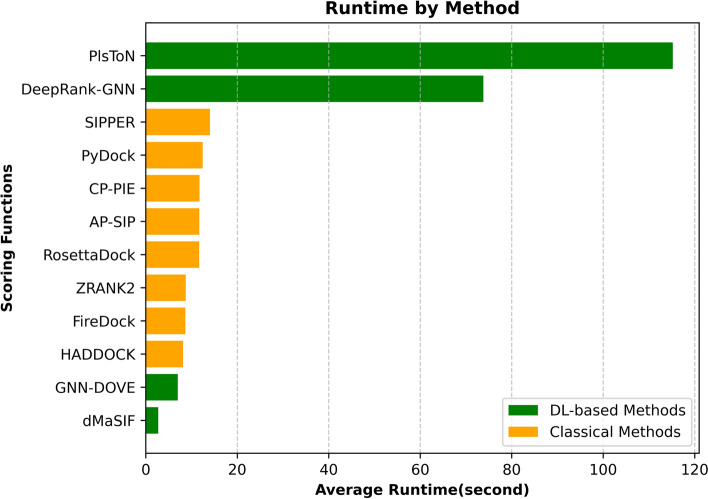
In this study, we perform a comprehensive survey of the state-of-the-art scoring functions by considering the most popular and highly performant approaches, both classical and deep learning-based, for scoring protein-protein complexes. The methods were also compared based on their runtime as it directly impacts their use in large-scale docking applications.
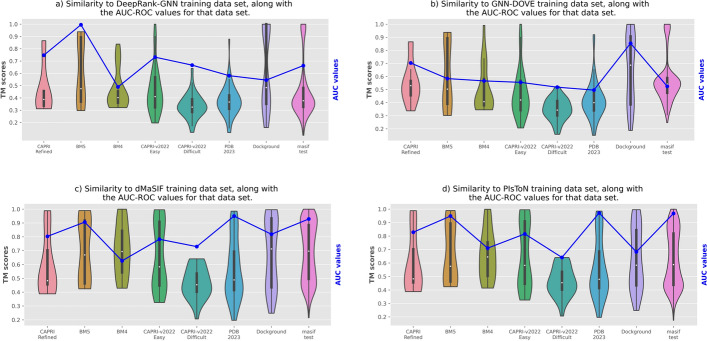
We evaluate the strengths and weaknesses of classical and deep learning-based approaches across seven public and popular datasets to aid researchers in understanding the progress made in this field.
虽然蛋白质-蛋白质对接对于我们理解蛋白质如何相互作用至关重要,但对蛋白质-蛋白质复合物构象进行评分是成功对接程序的关键组成部分。如果没有准确有效的评分函数来区分天然和非天然结合复合物,就无法保证当前对接工具的准确性。尽管已经提出了许多创新的评分函数,但用于对接的良好评分函数仍然难以捉摸。深度学习模型为使用明确的经验或数学函数对蛋白质-蛋白质复合物进行评分提供了替代方法。
在本研究中,我们通过考虑用于蛋白质-蛋白质复合物评分的最流行、性能最高的方法,对最先进的评分函数进行了全面调查,这些方法包括经典方法和基于深度学习的方法。还根据运行时间对这些方法进行了比较,因为运行时间直接影响它们在大规模对接应用中的使用。
我们在七个公共且流行的数据集上评估了经典方法和基于深度学习的方法的优缺点,以帮助研究人员了解该领域取得的进展。