Jeon Gye Sun, Nakamura Tomohiro, Lee Jeong-Seon, Choi Won-Jun, Ahn Suk-Won, Lee Kwang-Woo, Sung Jung-Joon, Lipton Stuart A
Department of Neurology, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Republic of Korea.
Mol Neurobiol. 2014 Apr;49(2):796-807. doi: 10.1007/s12035-013-8562-z. Epub 2013 Oct 4.
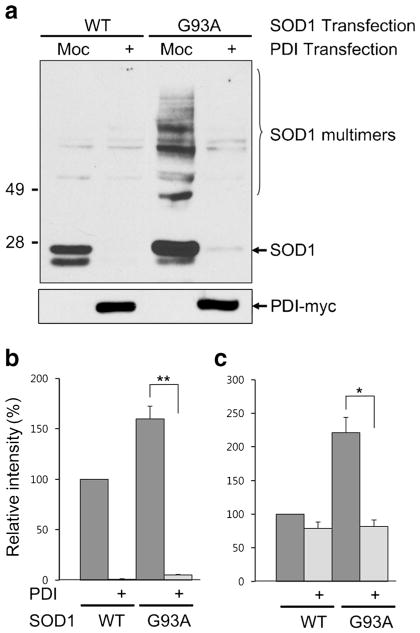
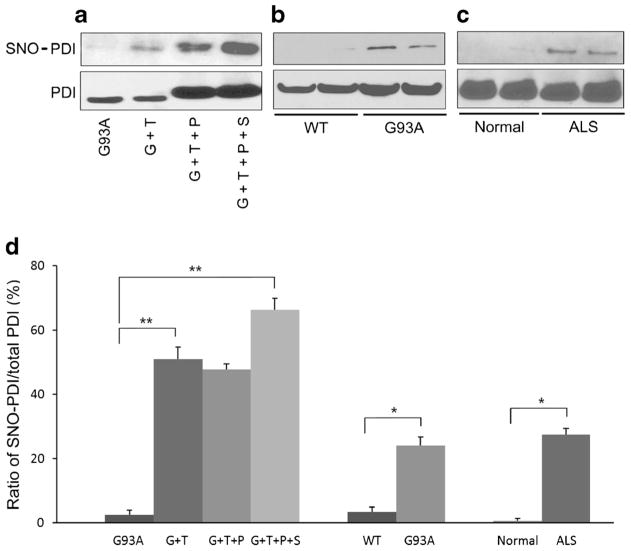
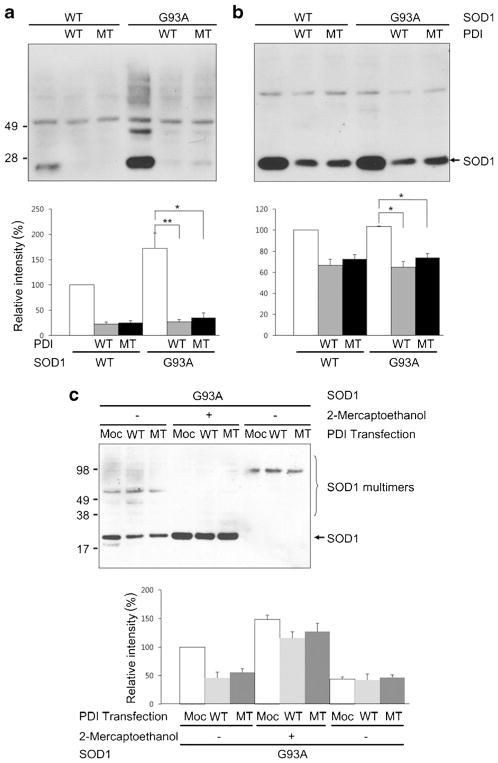
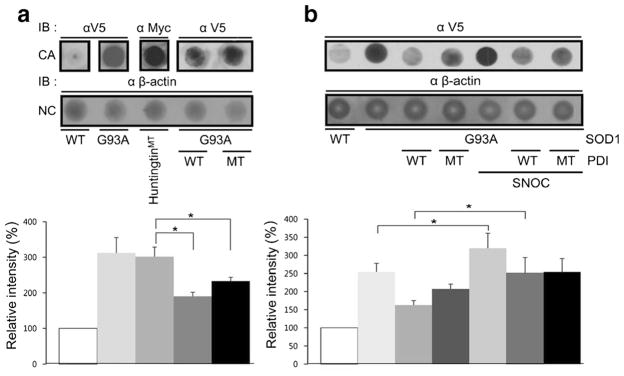
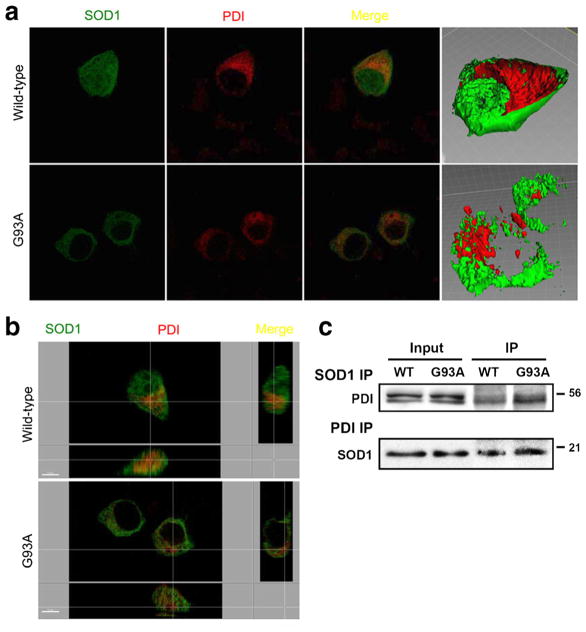
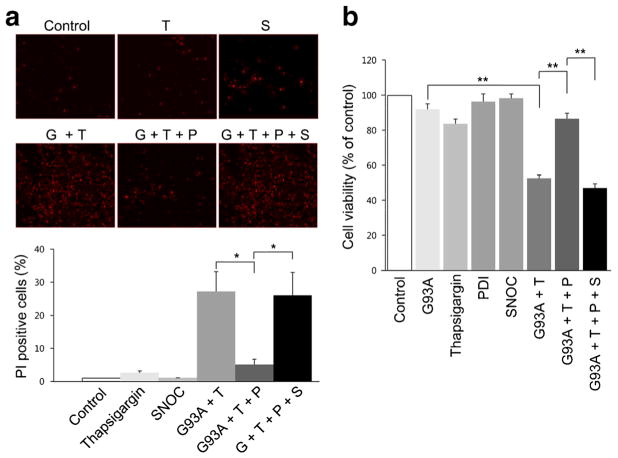
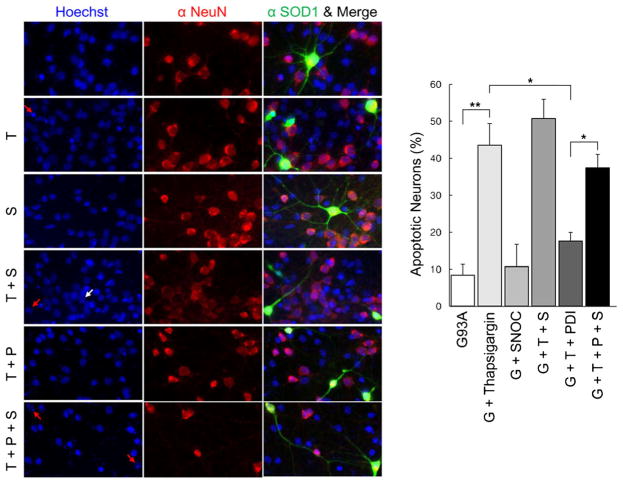
Aggregation of misfolded protein and resultant intracellular inclusion body formation are common hallmarks of mutant superoxide dismutase (mSOD1)-linked familial amyotrophic lateral sclerosis (FALS) and have been associated with the selective neuronal death. Protein disulfide isomerase (PDI) represents a family of enzymatic chaperones that can fold nascent and aberrant proteins in the endoplasmic reticulum (ER) lumen. Recently, our group found that S-nitrosylated PDI could contribute to protein misfolding and subsequent neuronal cell death. However, the exact role of PDI in the pathogenesis of ALS remains unclear. In this study, we propose that PDI attenuates aggregation of mutant/misfolded SOD1 and resultant neurotoxicity associated with ER stress. ER stress resulting in PDI dysfunction therefore provides a mechanistic link between deficits in molecular chaperones, accumulation of misfolded proteins, and neuronal death in neurodegenerative diseases. In contrast, S-nitrosylation of PDI inhibits its activity, increases mSOD1 aggregation, and increases neuronal cell death. Specifically, our data show that S-nitrosylation abrogates PDI-mediated attenuation of neuronal cell death triggered by thapsigargin. Biotin switch assays demonstrate S-nitrosylated PDI both in the spinal cords of SOD1 (G93A) mice and human patients with sporadic ALS. Therefore, denitrosylation of PDI may have therapeutic implications. Taken together, our results suggest a novel strategy involving PDI as a therapy to prevent mSOD1 aggregation and neuronal degeneration. Moreover, the data demonstrate that inactivation of PDI by S-nitrosylation occurs in both mSOD1-linked and sporadic forms of ALS in humans as well as mice.
错误折叠蛋白的聚集以及由此导致的细胞内包涵体形成是突变型超氧化物歧化酶(mSOD1)相关的家族性肌萎缩侧索硬化症(FALS)的常见特征,并与选择性神经元死亡有关。蛋白质二硫键异构酶(PDI)是一类酶伴侣蛋白家族,可在内质网(ER)腔中折叠新生的和异常的蛋白质。最近,我们小组发现S-亚硝基化的PDI可能导致蛋白质错误折叠并随后导致神经元细胞死亡。然而,PDI在肌萎缩侧索硬化症发病机制中的确切作用仍不清楚。在本研究中,我们提出PDI可减轻突变型/错误折叠的SOD1的聚集以及与内质网应激相关的神经毒性。导致PDI功能障碍的内质网应激因此在神经退行性疾病中分子伴侣缺陷、错误折叠蛋白积累和神经元死亡之间提供了一种机制联系。相反,PDI的S-亚硝基化会抑制其活性,增加mSOD1聚集,并增加神经元细胞死亡。具体而言,我们的数据表明S-亚硝基化消除了PDI介导的由毒胡萝卜素触发的神经元细胞死亡的减轻作用。生物素转换试验在SOD1(G93A)小鼠脊髓和散发性肌萎缩侧索硬化症人类患者中均检测到S-亚硝基化的PDI。因此,PDI的去亚硝基化可能具有治疗意义。综上所述,我们的结果提示了一种涉及将PDI作为预防mSOD1聚集和神经元变性的治疗方法的新策略。此外,数据表明在人类和小鼠的mSOD1相关型和散发性肌萎缩侧索硬化症中均发生了由S-亚硝基化导致的PDI失活。