School of Pharmacy & Centre for Biomolecular Sciences, University of Nottingham, University Park, Nottingham NG7 2RD, UK.
Bioorg Med Chem. 2013 Nov 15;21(22):6868-77. doi: 10.1016/j.bmc.2013.09.038. Epub 2013 Sep 25.
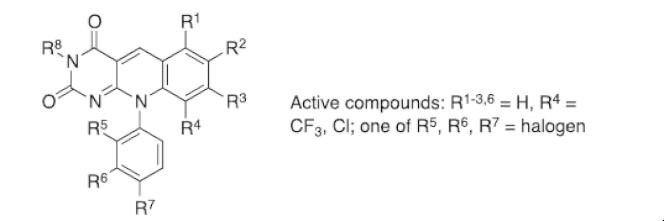
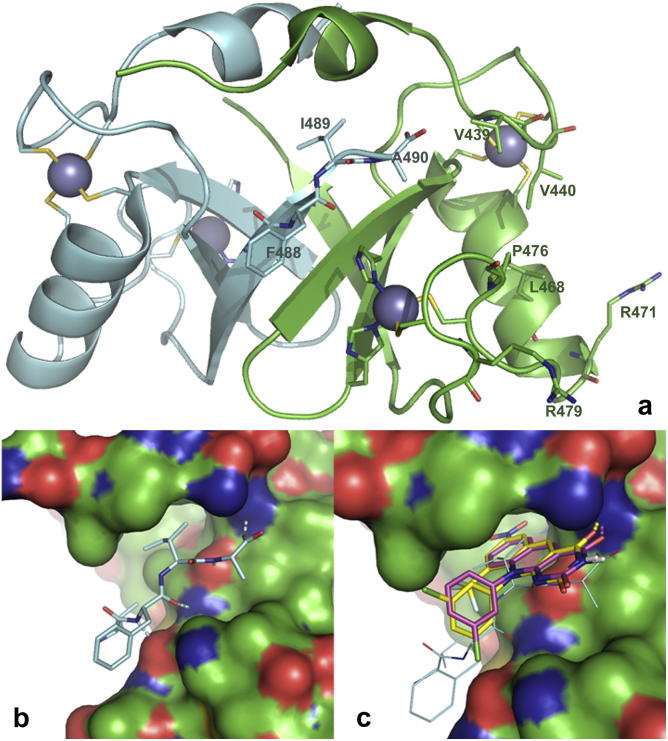
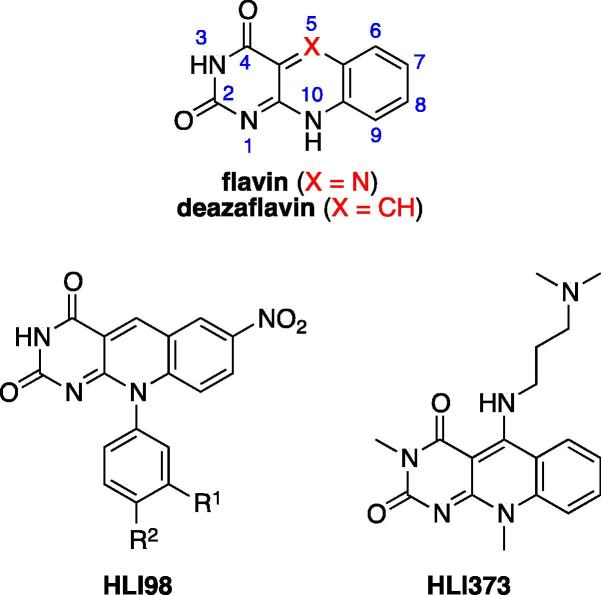
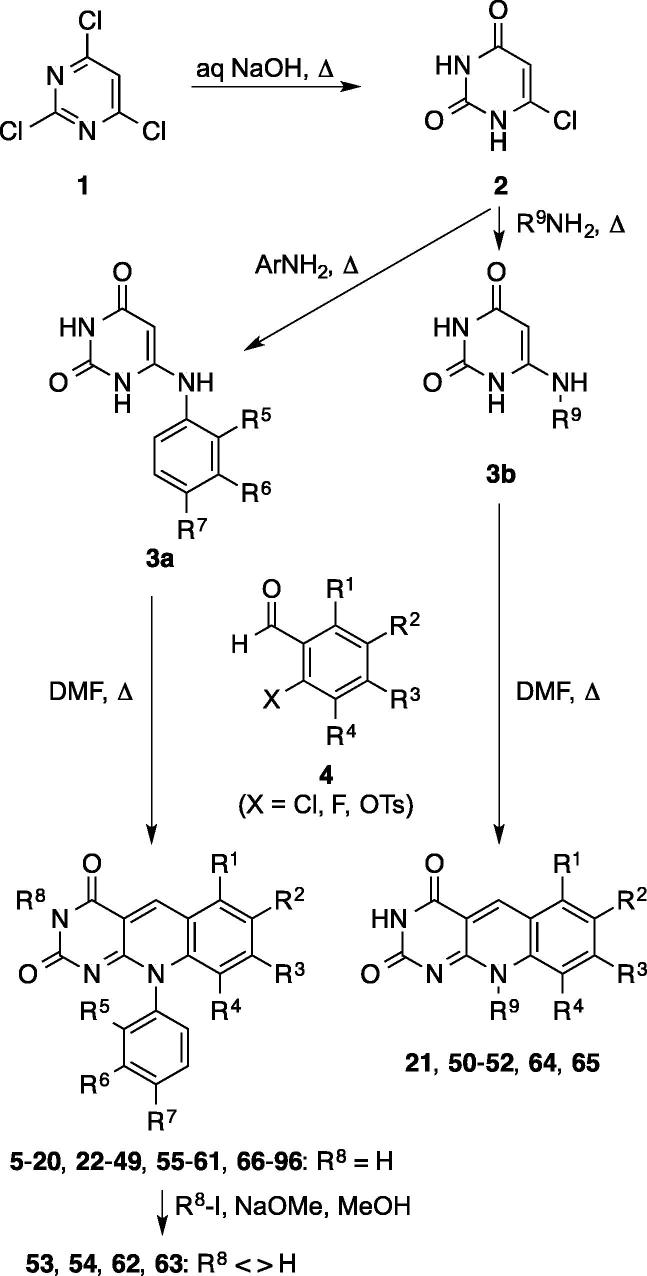
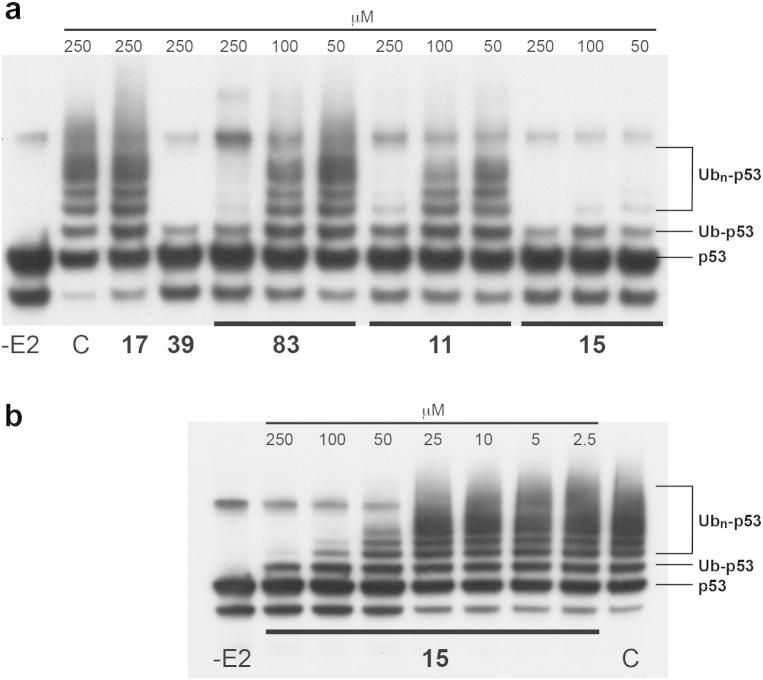
Based on previous reports of certain 5-deazaflavin derivatives being capable of activating the tumour suppressor p53 in cancer cells through inhibition of the p53-specific ubiquitin E3 ligase HDM2, we have conducted an structure-activity relationship (SAR) analysis through systematic modification of the 5-deazaflavin template. This analysis shows that HDM2-inhibitory activity depends on a combination of factors. The most active compounds (e.g., 15) contain a trifluoromethyl or chloro substituent at the deazaflavin C9 position and this activity depends to a large extent on the presence of at least one additional halogen or methyl substituent of the phenyl group at N10. Our SAR results, in combination with the HDM2 RING domain receptor recognition model we present, form the basis for the design of drug-like and potent activators of p53 for potential cancer therapy.
基于先前的报告,某些 5-脱氮黄素衍生物能够通过抑制 p53 特异性泛素 E3 连接酶 HDM2 来激活癌细胞中的肿瘤抑制因子 p53,我们通过对 5-脱氮黄素模板进行系统修饰进行了结构-活性关系(SAR)分析。该分析表明,HDM2 抑制活性取决于多种因素的组合。最活跃的化合物(例如 15)在去氮黄素 C9 位置含有三氟甲基或氯取代基,这种活性在很大程度上取决于 N10 位的苯环上至少还有一个额外的卤素或甲基取代基的存在。我们的 SAR 结果,结合我们提出的 HDM2 RING 结构域受体识别模型,为设计用于潜在癌症治疗的 p53 类药物和有效激活剂奠定了基础。