Bai Yongsheng, Cavalcoli James
Morgridge Institute for Research, University of Wisconsin-Madison, 330 N Orchard St, Madison, WI 53715, U.S.A.
Bioinformation. 2013 Oct 16;9(17):870-2. doi: 10.6026/97320630009870. eCollection 2013.
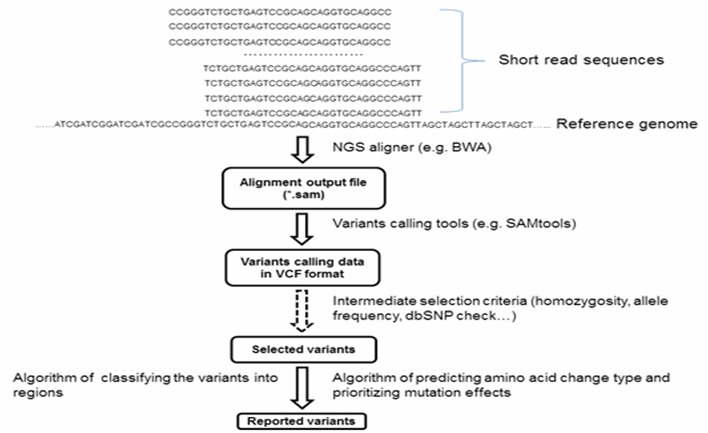
Many NGS analysis tools focusing on read alignment and variant calling functions for exome sequencing data have been developed in recent years. However, publicly available tools dealing with the downstream analysis of genome-wide variants are fewer and have limited functionality. We developed SNPAAMapper, a novel variant analysis pipeline that can effectively classify variants by region (e.g. CDS, UTRs, intron, upstream, downstream), predict amino acid change type (e.g. synonymous, non-synonymous mutation), and prioritize mutation effects (e.g. CDS versus UTRs). Additional functionality afforded by our pipeline includes: checking variants at exon/intron junctions, customized homozygosity and allele frequency cutoff parameters, and annotation of known variants with dbSNP information, listing original and mutated amino acid sequences containing variants. The final result is reported in a spreadsheet format table containing all variant associated information and prioritized amino acids effects for investigators to examine.
Perl scripts and required input files are available on the web at http://www.ccmb.med.umich.edu/ccdu /SNPAAMapper.
近年来,已经开发了许多专注于外显子测序数据的读段比对和变异检测功能的二代测序(NGS)分析工具。然而,处理全基因组变异下游分析的公开可用工具较少,且功能有限。我们开发了SNPAAMapper,这是一种新型变异分析流程,它可以按区域(例如编码序列(CDS)、非翻译区(UTRs)、内含子、上游、下游)有效分类变异,预测氨基酸变化类型(例如同义突变、非同义突变),并对突变效应进行优先级排序(例如CDS与UTRs)。我们的流程提供的其他功能包括:检查外显子/内含子交界处的变异、定制纯合性和等位基因频率截止参数,以及用dbSNP信息注释已知变异,列出包含变异的原始和突变氨基酸序列。最终结果以电子表格格式的表格报告,其中包含所有与变异相关的信息以及为研究人员检查而确定优先级的氨基酸效应。
Perl脚本和所需的输入文件可在网页http://www.ccmb.med.umich.edu/ccdu /SNPAAMapper上获取。