KTH Royal Institute of Technology, Science for Life Laboratory, School of Computer Science and Communication, Department of Computational Biology, Solna, Sweden.
BMC Bioinformatics. 2013 Nov 20;14:334. doi: 10.1186/1471-2105-14-334.
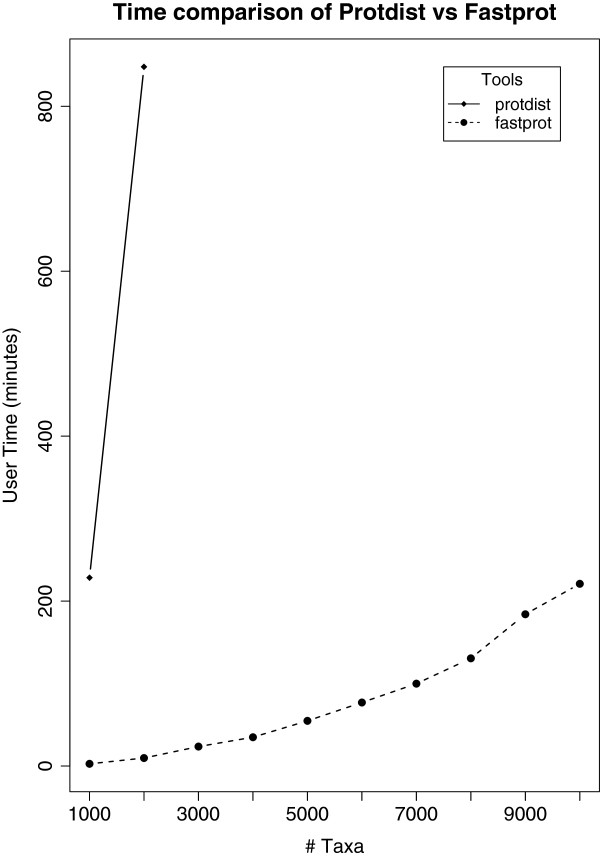
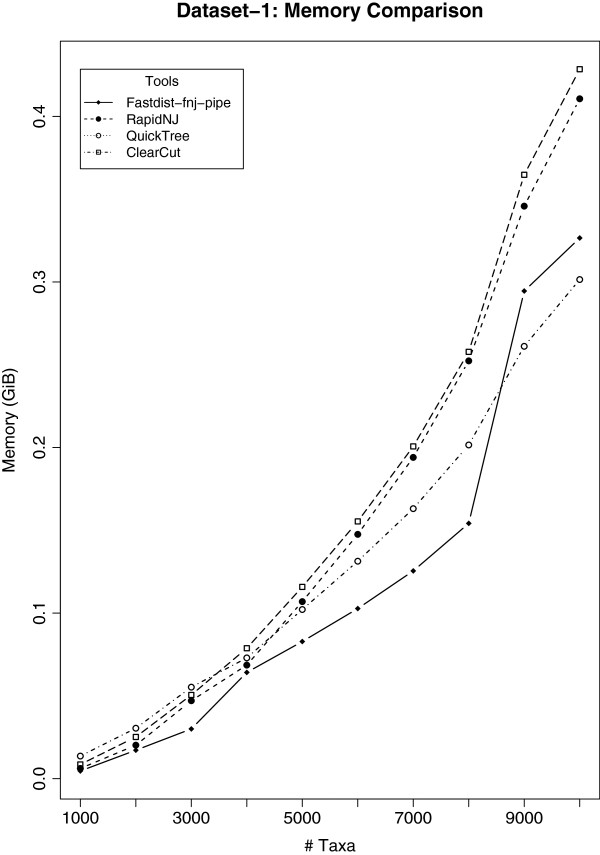
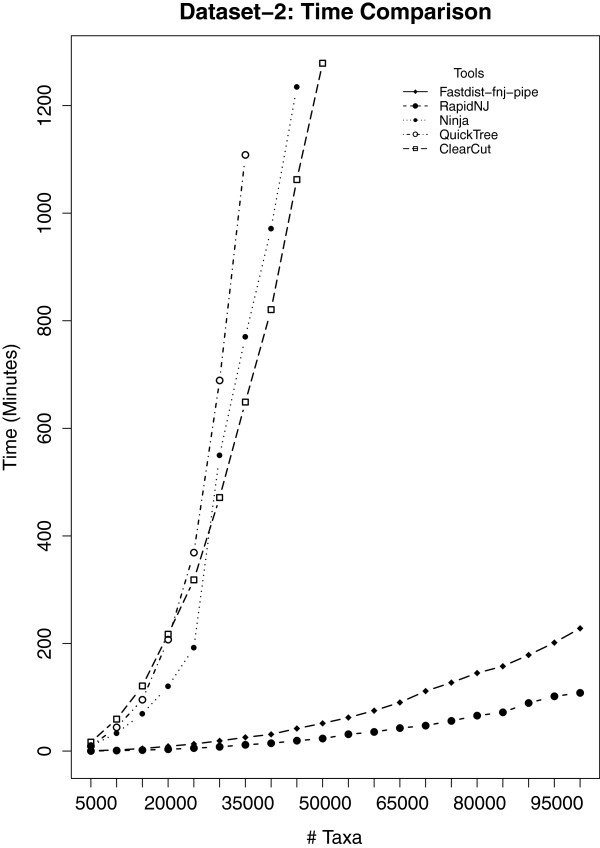
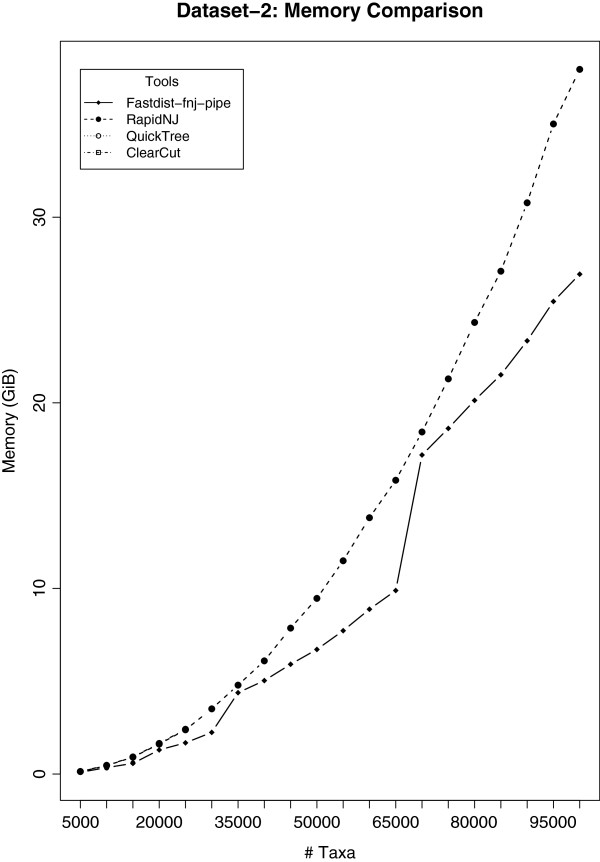
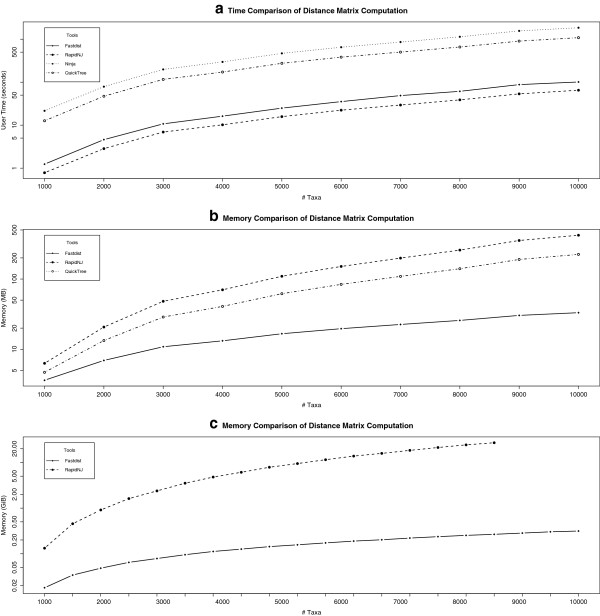
Distance methods are ubiquitous tools in phylogenetics. Their primary purpose may be to reconstruct evolutionary history, but they are also used as components in bioinformatic pipelines. However, poor computational efficiency has been a constraint on the applicability of distance methods on very large problem instances.
We present fastphylo, a software package containing implementations of efficient algorithms for two common problems in phylogenetics: estimating DNA/protein sequence distances and reconstructing a phylogeny from a distance matrix. We compare fastphylo with other neighbor joining based methods and report the results in terms of speed and memory efficiency.
Fastphylo is a fast, memory efficient, and easy to use software suite. Due to its modular architecture, fastphylo is a flexible tool for many phylogenetic studies.
距离方法是系统发生学中无处不在的工具。它们的主要目的可能是重建进化历史,但它们也被用作生物信息学管道中的组成部分。然而,计算效率低下一直是距离方法在非常大的问题实例上应用的限制。
我们提出了 fastphylo,这是一个软件包,其中包含了两种常见的系统发生学问题的高效算法的实现:估计 DNA/蛋白质序列距离和从距离矩阵重建系统发育。我们将 fastphylo 与其他基于邻接法的方法进行了比较,并根据速度和内存效率报告了结果。
Fastphylo 是一个快速、高效、易于使用的软件套件。由于其模块化架构,Fastphylo 是许多系统发育研究的灵活工具。