Department of Biology, University of Washington.
Genome Biol Evol. 2013;5(12):2410-9. doi: 10.1093/gbe/evt186.
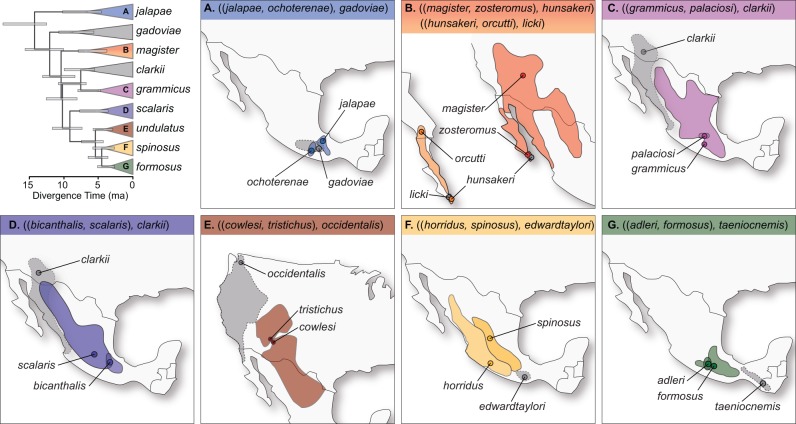
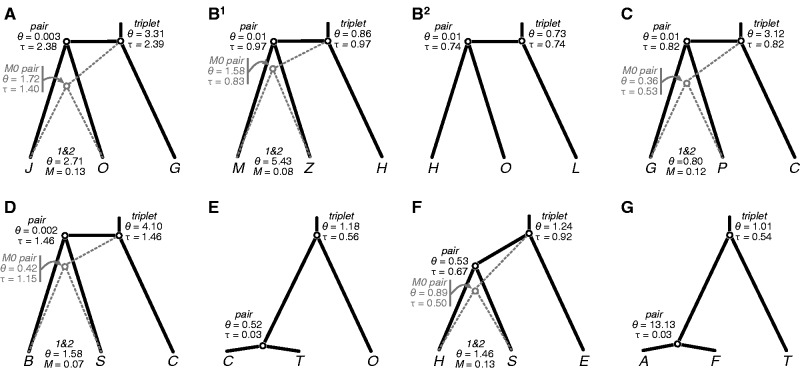
Species divergence is typically thought to occur in the absence of gene flow, but many empirical studies are discovering that gene flow may be more pervasive during species formation. Although many examples of divergence with gene flow have been identified, few clades have been investigated in a comparative manner, and fewer have been studied using genome-wide sequence data. We contrast species divergence genetic histories across eight triplets of North American Sceloporus lizards using a maximum likelihood implementation of the isolation-migration (IM) model. Gene flow at the time of species divergence is modeled indirectly as variation in species divergence time across the genome or explicitly using a migration rate parameter. Likelihood ratio tests (LRTs) are used to test the null model of no gene flow at speciation against these two alternative gene flow models. We also use the Akaike information criterion to rank the models. Hundreds of loci are needed for the LRTs to have statistical power, and we use genome sequencing of reduced representation libraries to obtain DNA sequence alignments at many loci (between 340 and 3,478; mean = 1,678) for each triplet. We find that current species distributions are a poor predictor of whether a species pair diverged with gene flow. Interrogating the genome using the triplet method expedites the comparative study of species divergence history and the estimation of genetic parameters associated with speciation.
物种分化通常被认为是在没有基因流动的情况下发生的,但许多实证研究发现,基因流动在物种形成过程中可能更为普遍。尽管已经确定了许多有基因流动的分化例子,但很少有类群以比较的方式进行研究,也很少有使用全基因组序列数据进行研究。我们使用隔离-迁移(IM)模型的最大似然实现,对比了北美洲石龙子 8 对的物种分化遗传历史。物种分化时的基因流动通过在整个基因组上的物种分化时间的变化或使用迁移率参数来间接建模。似然比检验(LRT)用于检验无基因流动的零模型与这两种替代基因流动模型的分歧。我们还使用赤池信息量准则对模型进行排名。LRT 需要数百个基因座才能具有统计学效力,我们使用简化代表性文库的基因组测序来获得每个三联体的许多基因座(340 到 3478 个之间;平均值为 1678 个)的 DNA 序列比对。我们发现,目前的物种分布是预测物种对是否有基因流动分化的一个很差的指标。使用三联体方法研究基因组可以加速对物种分化历史的比较研究和与物种形成相关的遗传参数的估计。