Division of Oncology, Stanford University School of Medicine, Stanford, CA 94305, USA.
BMC Med Genomics. 2013 Dec 5;6:54. doi: 10.1186/1755-8794-6-54.
Colorectal cancer is the third leading cause of cancer deaths in the United States. The initial assessment of colorectal cancer involves clinical staging that takes into account the extent of primary tumor invasion, determining the number of lymph nodes with metastatic cancer and the identification of metastatic sites in other organs. Advanced clinical stage indicates metastatic cancer, either in regional lymph nodes or in distant organs. While the genomic and genetic basis of colorectal cancer has been elucidated to some degree, less is known about the identity of specific cancer genes that are associated with advanced clinical stage and metastasis.
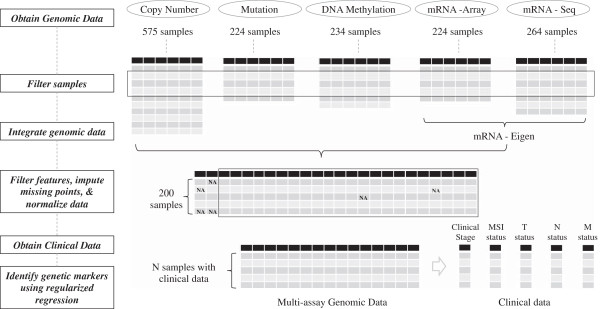
We compiled multiple genomic data types (mutations, copy number alterations, gene expression and methylation status) as well as clinical meta-data from The Cancer Genome Atlas (TCGA). We used an elastic-net regularized regression method on the combined genomic data to identify genetic aberrations and their associated cancer genes that are indicators of clinical stage. We ranked candidate genes by their regression coefficient and level of support from multiple assay modalities.
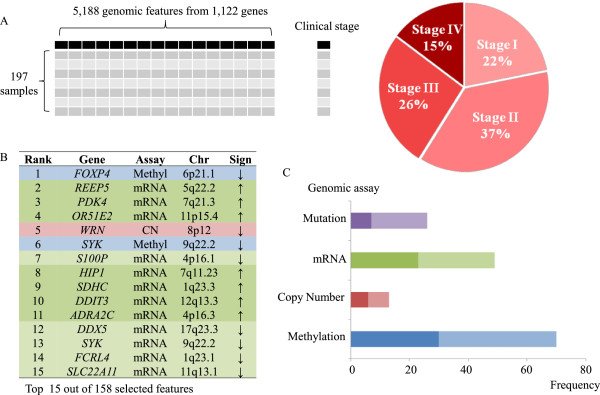
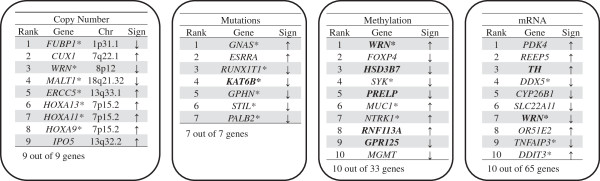
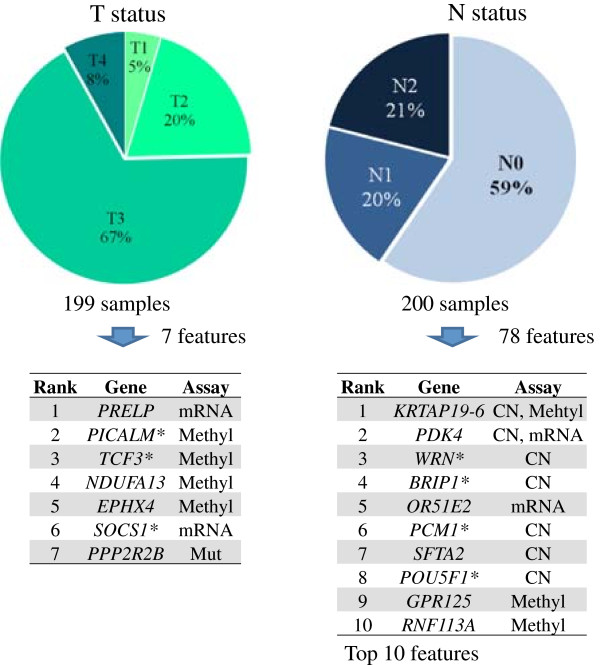
A fit of the elastic-net regularized regression to 197 samples and integrated analysis of four genomic platforms identified the set of top gene predictors of advanced clinical stage, including: WRN, SYK, DDX5 and ADRA2C. These genetic features were identified robustly in bootstrap resampling analysis.
We conducted an analysis integrating multiple genomic features including mutations, copy number alterations, gene expression and methylation. This integrated approach in which one considers all of these genomic features performs better than any individual genomic assay. We identified multiple genes that robustly delineate advanced clinical stage, suggesting their possible role in colorectal cancer metastatic progression.
结直肠癌是美国癌症死亡的第三大主要原因。结直肠癌的初步评估包括临床分期,考虑原发肿瘤侵袭的程度,确定有转移性癌症的淋巴结数量以及确定其他器官的转移部位。晚期临床分期表明存在局部淋巴结或远处器官的转移性癌症。尽管已经在一定程度上阐明了结直肠癌的基因组和遗传基础,但对于与晚期临床分期和转移相关的特定癌症基因的身份了解较少。
我们从癌症基因组图谱(TCGA)中编译了多种基因组数据类型(突变,拷贝数改变,基因表达和甲基化状态)以及临床元数据。我们使用结合基因组数据的弹性网络正则化回归方法来识别遗传异常及其相关的癌症基因,这些异常和基因是临床分期的指标。我们根据回归系数和来自多种检测方式的支持程度对候选基因进行排名。
对 197 个样本进行弹性网络正则化回归拟合以及对四个基因组平台进行综合分析,确定了一组高级临床分期的顶级基因预测因子,包括:WRN、SYK、DDX5 和 ADRA2C。在自举重采样分析中,这些遗传特征被稳健地识别。
我们进行了一项整合多种基因组特征的分析,包括突变、拷贝数改变、基因表达和甲基化。这种综合方法考虑了所有这些基因组特征,其性能优于任何单个基因组检测。我们确定了多个基因,这些基因可以明确区分晚期临床分期,这表明它们可能在结直肠癌转移进展中发挥作用。