Structural Genomics Consortium, Nuffield Department of Medicine, University of Oxford , Old Road Campus Research Building, Roosevelt Drive, Oxford OX3 7DQ, U.K.
J Med Chem. 2014 Jan 23;57(2):462-76. doi: 10.1021/jm401568s. Epub 2013 Dec 30.
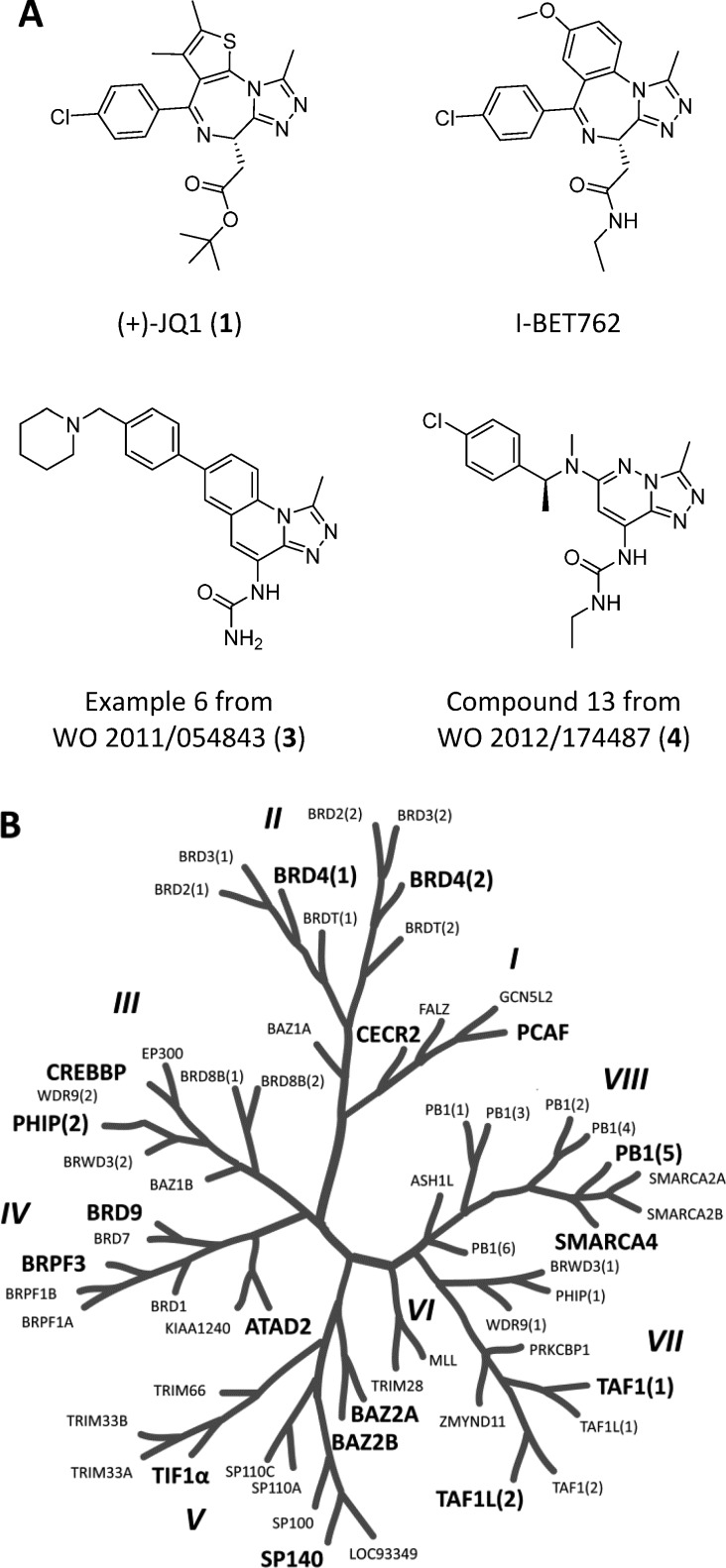
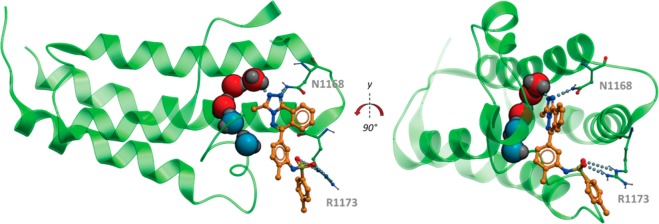
Bromodomains are gaining increasing interest as drug targets. Commercially sourced and de novo synthesized substituted [1,2,4]triazolo[4,3-a]phthalazines are potent inhibitors of both the BET bromodomains such as BRD4 as well as bromodomains outside the BET family such as BRD9, CECR2, and CREBBP. This new series of compounds is the first example of submicromolar inhibitors of bromodomains outside the BET subfamily. Representative compounds are active in cells exhibiting potent cellular inhibition activity in a FRAP model of CREBBP and chromatin association. The compounds described are valuable starting points for discovery of selective bromodomain inhibitors and inhibitors with mixed bromodomain pharmacology.
溴结构域作为药物靶点正受到越来越多的关注。商业来源和从头合成的取代 [1,2,4]三唑并[4,3-a]并[1,2,5]噻二嗪是 BET 溴结构域(如 BRD4)以及 BET 家族以外的溴结构域(如 BRD9、CECR2 和 CREBBP)的强效抑制剂。这一系列新化合物是 BET 亚家族以外的溴结构域的首个亚微摩尔抑制剂实例。代表性化合物在 CREBBP 的 FRAP 模型和染色质结合中表现出强效的细胞抑制活性,在细胞中具有活性。所描述的化合物是发现选择性溴结构域抑制剂和具有混合溴结构域药理学的抑制剂的有价值的起点。