Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
J Neuroinflammation. 2013 Dec 5;10:146. doi: 10.1186/1742-2094-10-146.
Mutations in proteolipid protein (PLP), the most abundant myelin protein in the CNS, cause the X-linked dysmyelinating leukodystrophies, Pelizaeus-Merzbacher disease (PMD) and spastic paraplegia type 2 (SPG2). Point mutations, deletion, and duplication of the PLP1 gene cause PMD/SPG2 with varying clinical presentation. Deletion of an intronic splicing enhancer (ISEdel) within intron 3 of the PLP1 gene is associated with a mild form of PMD. Clinical and preclinical studies have indicated that mutations in myelin proteins, including PLP, can induce neuroinflammation, but the temporal and spatial onset of the reactive glia response in a clinically relevant mild form of PMD has not been defined.
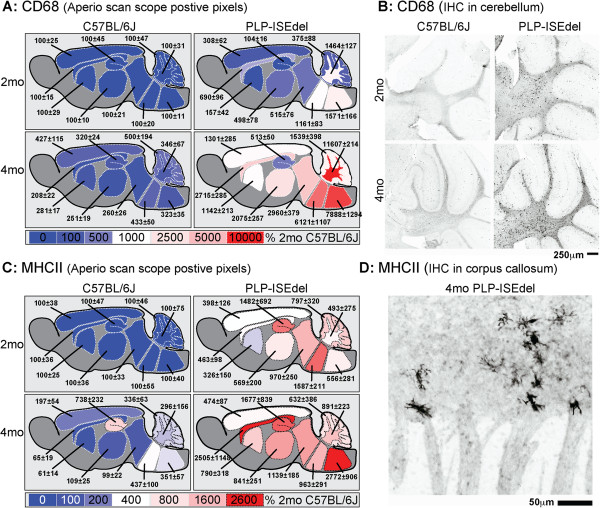
A PLP-ISEdel knockin mouse was used to examine the behavioral and neuroinflammatory consequences of a deletion within intron 3 of the PLP gene, at two time points (two and four months old) early in the pathological progression. Mice were characterized functionally using the open field task, elevated plus maze, and nesting behavior. Quantitative neuropathological analysis was for markers of astrocytes (GFAP), microglia (IBA1, CD68, MHCII) and axons (APP). The Aperio ScanScope was used to generate a digital, high magnification photomicrograph of entire brain sections. These digital slides were used to quantify the immunohistochemical staining in ten different brain regions to assess the regional heterogeneity in the reactive astrocyte and microglial response.
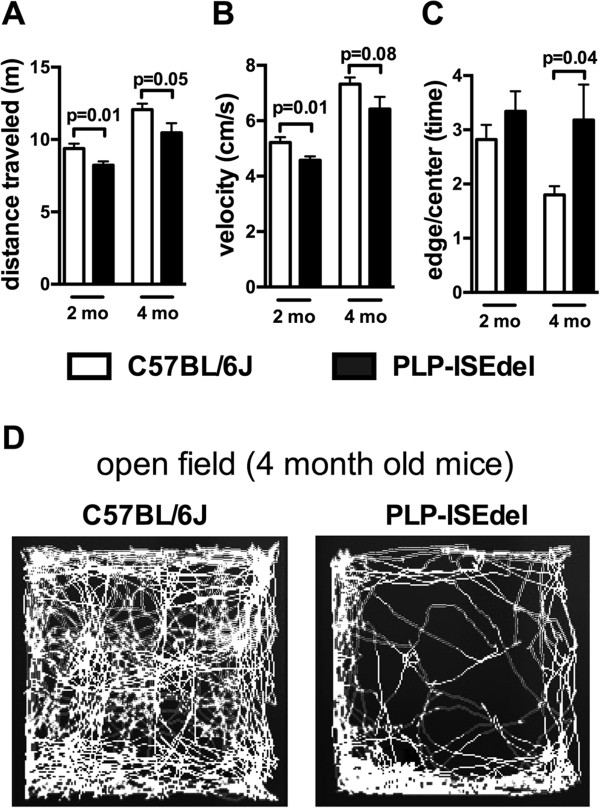
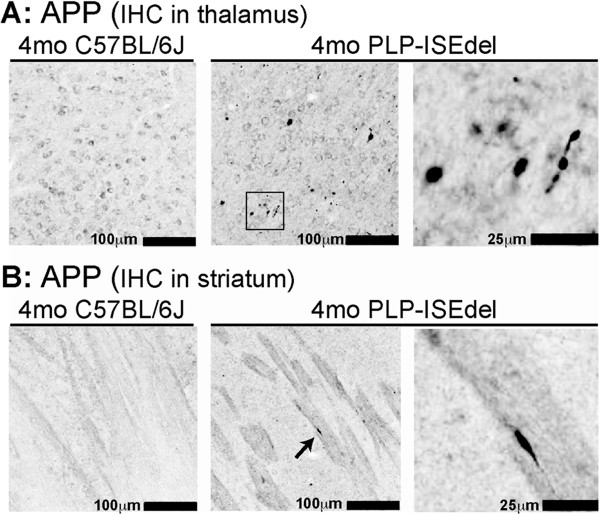
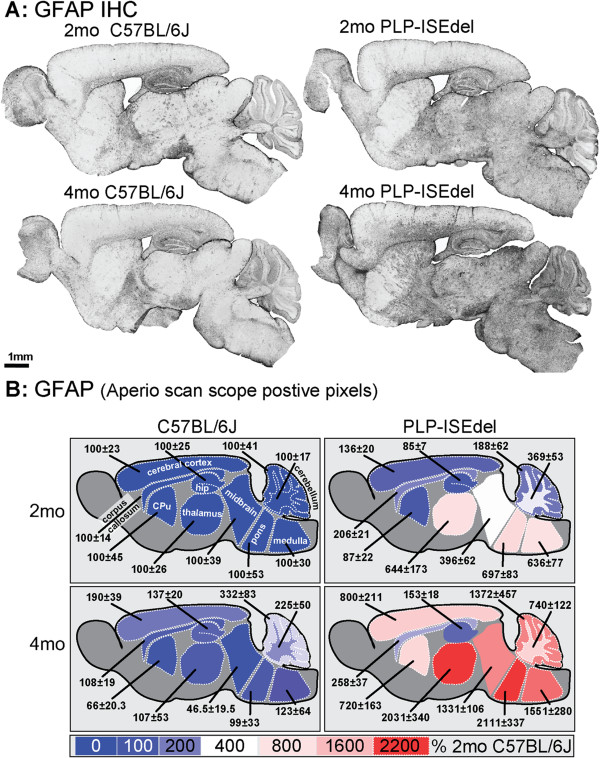
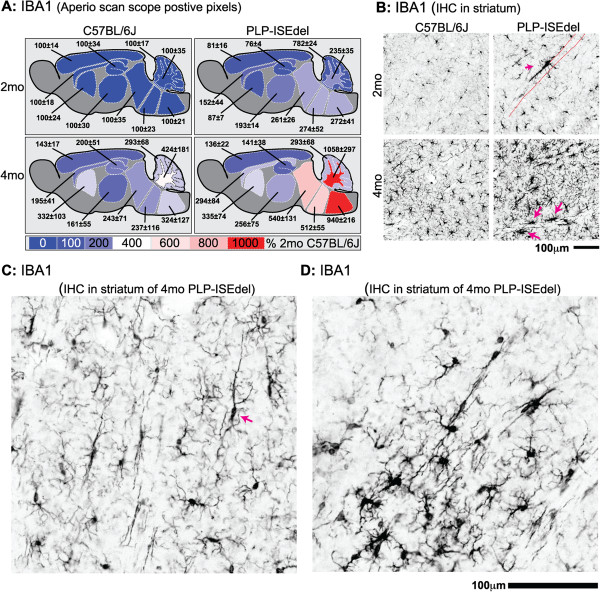
The PLP-ISEdel mice exhibited behavioral deficits in the open field and nesting behavior at two months, which did not worsen by four months of age. A marker of axonal injury (APP) increased from two months to four months of age. Striking was the robust reactive astrocyte and microglia response which was also progressive. In the two-month-old mice, the astrocyte and microglia reactivity was most apparent in white matter rich regions of the brain. By four months of age the gliosis had become widespread and included both white as well as gray matter regions of the brain.
Our results indicate, along with other preclinical models of PMD, that an early reactive glia response occurs following mutations in the PLP gene, which may represent a potentially clinically relevant, oligodendrocyte-independent therapeutic target for PMD.
在中枢神经系统中,髓鞘蛋白脂蛋白(PLP)的突变导致 X 连锁的脱髓鞘性脑白质营养不良,佩利兹默布赫尔病(PMD)和痉挛性截瘫 2 型(SPG2)。PLP1 基因突变、缺失和重复导致 PMD/SPG2,临床表现不同。PLP1 基因内含子 3 内内含子剪接增强子(ISEdel)的缺失与 PMD 的轻度形式有关。临床和临床前研究表明,髓鞘蛋白(包括 PLP)的突变可诱导神经炎症,但在临床上相关的 PMD 轻度形式中,反应性神经胶质反应的时空起始尚未确定。
使用 PLP-ISEdel 基因敲入小鼠,在疾病早期(两个月和四个月大),检查 PLP 基因内含子 3 内缺失对神经炎症的影响。使用开放场任务、高架十字迷宫和筑巢行为对小鼠进行功能特征描述。定量神经病理学分析用于星形胶质细胞(GFAP)、小胶质细胞(IBA1、CD68、MHCII)和轴突(APP)的标志物。Aperio ScanScope 用于生成整个脑切片的数字、高倍显微镜照片。这些数字幻灯片用于定量评估十个不同脑区的免疫组织化学染色,以评估反应性星形胶质细胞和小胶质细胞反应的区域异质性。
PLP-ISEdel 小鼠在两个月大时表现出开放场和筑巢行为的缺陷,四个月大时没有恶化。轴突损伤标志物(APP)从两个月增加到四个月。引人注目的是强烈的反应性星形胶质细胞和小胶质细胞反应,且呈进行性。在两个月大的小鼠中,星形胶质细胞和小胶质细胞的反应最明显的是富含白质的脑区。四个月大时,神经胶质增生已广泛存在,包括脑的白质和灰质区域。
我们的结果表明,与 PMD 的其他临床前模型一样,在 PLP 基因突变后,会发生早期的反应性神经胶质反应,这可能代表 PMD 的一个潜在的、临床上相关的、少突胶质细胞非依赖性治疗靶点。